seqtools revision history

|
On my Windows 7 pro pc seqtools seems to install and function without any problems. Feedback from other users on this issue would be highly appreciated... |
| I couldn't resist including this very nice animated picture of two of my favourite actors reminding us of the remarkable results of complex gene expression... |
revision history
|
8.4.066 - 16.09.2010 - Seqtools Configuration. - Minor correction of the automatic update function
for seqtools.exe available on the
Help/SEQtools Configuration.../ form.
This update requires a new download and install of Seqtools to be effective. |
|
8.4.050 - 20.08.2010 - QBlast function for batch blast search is repaired. - As a result of slight modifications of the QBlast server at NCBI the
QBlast function in seqtools has not been working for a while. The code in this build of seqtools has been
adjusted to handle these changes - and the function for batch blast search on the QBlast server now seems
to work. |
|
8.4.045 - 27.03.2010 - A few - unimportant - adjustments of form layouts. - Somewhat to my surprise, the number of
seqtools users is slowly but steadily increasing. Now being retired, I seldom use the program myself
execpt for occasional testing of various functions included in seqtools. But I am pleased that other
persons still find my program suite useful in their struggle with dna and protein sequences. |
|
8.4.042 - 06.09.2008 - Bug in local blast search functions corrected. - Two quite serious bugs recently detected by Birgitt Oeser have been corrected in this version.
|
|
8.4.040 - 17.06.2008 - New option added to header search function. - In response to a request by Patrick Gaffey an option has been added to
the header search function. It is now possible to step through matching strings on the displayed header page by repeatedly clicking the "Find Next" command button on the small form handling header search results. |
|
8.4.032 - 15.01.2008 - New facility added to the Clustering function - In response to the request of Julien Sacr� I have added options to remove from the current project either the sequences included in clusters or the unique sequences not included in clusters from the current project. The sequences are NOT deleted from the disk, just removed from the project. |
|
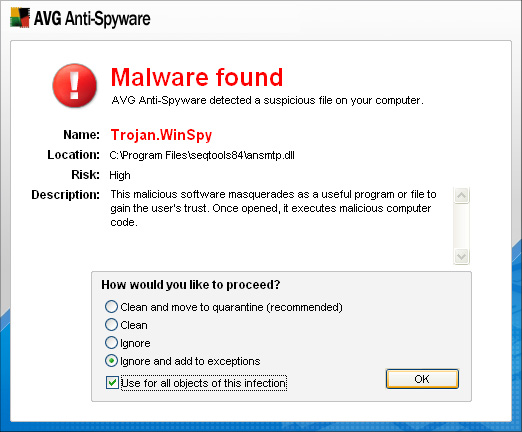
False positive virus / spyware warning - 11.11.2007 - There has
been a few reports from users that a file included in the seqtools setup is a trojan: Trojan.WinSpy as shown in the AVG warning below. Cheers |
|
What is your opinion about seqtools? - 13.05.2007 - As the author of seqtools I often
speculate how users receive and use the program. I have invested a long time writing and supporting the
program constantly trying to make the program as user friendly as possible. |
|
8.4.020 - 12.05.2007 - Virus problems with ANSMTP.dll - I have recently received two mails from users who reported
that they got a warning from their antivirus progams (in one case: Avast Home Edition) telling them that the
ansmtp.dll file is a virus. |
|
8.4.020 - 06.05.2007 - Major revision - I have received some user complaints after the release of the previous
"upgrade" which deserve some comments and explanations. The main reason for a series of problems was a note from
a user who mentioned that he no longer was able to load multi-sequence files generated with pervious versions of
seqtools. At this time I had just routinely updated the NCBI programs - which I usually do, and have done for several
years - when new releases become available. |
|
8.4.016 - 26.03.2007 - Major revision - This revision at the same time terminates the old seqtools 8.2 as well as the
separate 8.3 version for teaching purposes. In the future only one version - 8.4 - of seqtools will be available. |
|
8.3.052 - 13.10.2006 - Bug fix - Problems associated with closing the seqtools application have been fixed. This correction has also been implemented in seqtools version 8.2.108 for students. |
|
8.3.040 - 26.01.2006 - Bug fixes - (1) The active codon usage tabel can now be set and changed under the Preferences menu. (2) Calculating protein properties now also works when amino acid sequences contain lower-case letters. (3) A new routine has been implemented to handle e-mail based operations. This affects the bug-reporting facility under the Help menu and the sequence forwarding facility under the File menu. |
|
8.3.037 - 11.12.2005 - A new "school edition" of Seqtools released
- Several users have previously been employing Seqtools in elementary and high school teaching as well as in undergraduate university courses in bioinformatics. As the large number of functions and facilities in Seqtools often leads to confusion, a special
version of Seqtools have been released. |
|
8.3.033 - 23.10.2005 - The revision of the web manual is finally completed
- During this revision
all routines and functions in SEQtools were tested and corrected where necessary making this version of SEQtools
probably the most stable and bug-free. |
|
8.3.030 - 22.09.2005 - A serious bug in the batch blast function corrected
- Thanks to Dag H. Coucheron a serious bug was detected which caused the parser for returned
blast results from genbank/NCBI to ignore all description lines in the results file.
This bug is corrected now and so is a number of less important bugs here and there. More
bugs will probably be found as the manual revision/program testing progresses. So please be
patient - and send me a note when/if you detect something weird. |
|
8.3.025 - 12.09.2005 - Trim raw sequence trace files rewritten...
- This function suffered from several bugs - one preventing it from running under Windows XP operating systems. These bugs have been corrected (I hope...) and the functions now do what they were intended to do. |
|
8.3.021 - 30.05.2005 - Options to export sequence list as Excel spreadsheet... - This function has been a wish of many users for a long time. On my own pc it works perfectly well. However, I am not sure how the update and installation files handle the setup of the Excel part. Please let me know if you experience any problems updating seqtools to this build. In addition to this function, a number of improvements is included in this update, the most important being a greatly increased stability of the batch blast search function. Recently almost 17,000 protein sequences were searched against genbank "nr" without any user intervention at all. The search took several days. |
corrections to version 8.2
|
8.2.098 - 08.12.2004 - Several functions modified to increase speed... - The dot-matrix sequence comparison function has been improved to improve speed. The function is, howver, still not among the fastest, especially when sequence length exceeds some thousand bases. The sequence complementing function is also slightly improved. This version of the executable is compiled as so-called p-code which according to several experts should generate faster code and - as an extra bonus - also results in a considerably smaller exe-file. If you experience any problems please let me know. 8.2.095-97 - 22.11.2004 - Clustering output forms redesigned... - The design of the forms used to display the results of sequence clustering has long been rather unsatisfactory. This has been changed in this update. The resulting clusters are now listed in a tree-view control which links directly to the form displaying members of the selected cluster. Clicking the name of a cluster in the tree lists highlights all members of the cluster on the group form for easy selection of cluster members for alignment and/or merging. Clicking a single member of a cluster in the tree also lists the entire cluster but only highlights the selected sequence on the group form. As before, holding down the right mouse button on the group form displays the blast description lines for selected cluster member - provided of course that blast results are available. The previous report form is now only used for printing and saving a summary of the clustering. The report form is opened from the main clustering form under the report menu. The print and save options are contained under the file menu of the report form. Minor changes in this version includes improved setting of the number of descriptions, alignments and expect value cutoffs for blast searches on all blast search forms. Finally, note that the collection of auxiliary components has been updated again. This time because of a recent upgrade of the chromatogram processing program from the Sanger Institute used by seqtools. I have included a new option on the download page of the web which allows you to download the core seqtools installation and the auxiliary and emboss components as a single installation file. It is, however, still possible to update each component individually from the seqtools configuration form. 8.2.094 - 14.11.2004 - Bugs in clustering function corrected... - A bug preventing clustering large number of sequences has been corrected. Problems with loading and saving (with and without compression) large multi-sequence files have been corrected. 8.2.092 - 26.10.2004 - Bugs in save/export function corrected... - A bug preventing saving / exporting project data has been corrected. 8.2.091 - 04.10.2004 - Bugs in contig builder function, Windows bug!!...
-
A bug preventing selection of sequences to be merged is corrected. A new menu item has been added
to the File menu enabeling you to close the current project without exiting seqtools.
8.2.085 - 20.08.2004 - Several minor bugs corrected... - Small changes here and there. Rename functions with more readable sequence name lists. Clicking the Exit key on the Registration form no longer causes a crash of seqtools. 8.2.083 - 23.07.2004 - Yet another attempt to improve auto-updating seqtools... It has been unexpectedly difficult to get the program update facility to work properly due to differences in operating systems, server setup, firewalls etc. Several users have complained that seqtools freezes instead of downloading the update. This build addresses this problem. 8.2.082 - 20.07.2004 - New option for editing clusters... - It is now possible to display and edit the individual members of a cluster after clustering thanks to the suggestion by Florent Crepineau. Remember to let me know if you need functions or facilities not already included in seqtools. Often it is quite simple to modify seqtools to precisely match your needs. 8.2.080 - 08.07.2004 - Bugs in trimming raw sequences... - Several minor bugs in the function for trimming raw sequences under the Edit menu have been corrected in this build. User feed-back in general and related to this function in particular are highly welcome. 8.2.075 23.06.2004 - Bug in checking for an active IN connection... - It appears that some users still experience problems with the function testing if a live internet connection is available. This apparently trivial test has caused a lot of troubles in part due to bugs in the microsoft api function which behaves differently with different versions of windows (XP and win2000). When I test the function on my own PC's everything works as it should. Version 8.2.075 includes a second user option: "Ignore" which cancels internet connection checking by assuming that a live connection is available. 8.2.071 17.05.2004 - Bug in checking for an active IN connection...
- Recent versions of seqtools contained a bug in the routine checking the internet connection
status of the pc. The user was informed that no internet connection was present irrespectively
of the connection state of the pc. This problem is corrected in version 8.2.071. Registered user
have been informed in a recent newsletter. Other users should upgrade to version 8.2.071. 8.2.051 24.03.2004 - More corrections...
- Thanks to several user who have pointed out a few bugs and misbehaviours of seqtools this version,
8.2.051 may contain fewer bugs. 10.03.2004 - Various corrections to several functions
- Corrections have been made to many functions including: Blast blatch searching, description line
formatting, management of information stored in sequence headers, facility for entering user comments,
project save/export function. New functions include options to retrieve information from header
description line sections generated with local database searches. 30.01.2004 - QBlast batch blast function
- I failed to realise that the maximum string length for an url in Microsoft IE is 2083 characters.
As the first edition of the qblast function in seqtools used this method to submit blast requests to
NCBI this resulted in submission failure if sequence length exceded some 1500 bases - which is
obviously not satisfactory. 25.01.2004 - Menu icons added to selected forms
- With the large number of functions and options in SEQtools it is sometimes difficult to locate a
specific facility without having to read several menu item captions. Therefore I have added various
icons to menu items on the main editor, the header form and the sequence listing form in version 8.2. |
corrections to version 8.1
|
06.01.2004 - Various corrections to 8.1
- A new function has been added for batch blast searching at NCBI. The new function is built on the
urlapi (QBlast) made available by NCBI
and seems to work quite well. The new function circumvents firewall related problems, is quite fast
and hopefully do not suffer from the difficulties of the Blastcl3 based old function. The two versions
of batch blast search functions will co-exist for a while in seqtools until the QBlast function has
been thoroughly tested. 04.12.2003 - Various corrections to 8.1 - Several bugs have been corrected and various improvements made in the following functions: EST submission, loading fasta files, fasta definition line editor, blast sequential search, loading and saving project files, loading ms files via the commandline, trimming polyA tails. 05.10.2003 - Various corrections to 8.1
- Several bugs in the EST submission function, Header compose function, Loading of fasta formatted
multi-sequence files, Fasta definition line editor. Speed improved by various fine-tuning the code.
Please continue reporting bugs in the new version... 03.10.2003 - Various corrections to 8.1
- Several bugs removed and code improved in: 1) Loading of multi-sequence files, 2) Multiple local
database blast search and tabular listing of results, 3) Header composition function, 4) Fasta definition
line editor, 5) Manual installation of auxiliary and emboss programs, 6) SEQtools installed components
listing, 7) Save/Export sequences. 02.10.2003 - Version 8.1 of seqtools released
- Handling of information stored in sequence headers has been completely redesigned in version 8.1. The
header compose form now gives you complete freedom to display and list any type of information
contained in headers. With a few simple checkmarks you can select one or more blast search results
plus a number of other information types such as genbank records and medline entries, various types
of imported information, local multi-database searches etc. |
corrections to version 8.0
|
29.09.2003 - Save and Export function merged, Sequential Batch Blast function modified
- The Save Sequence and the Sequence Export forms have been merged into a single save form which
includes the combined functionality of both functions. 25.09.2003 - New sequential batch blast function
- Sometimes a blast search returns a result which fails to reveal a putative function of the gene
returned by the search. To improve this situation a new batch blast function has been added to
SEQtools which allows you to perform sequential batch blast searches. 04.09.2003 - Various improvements
- A sequence list field has been added to the main header form making navigation between headers
more convenient. 25.08.2003 - Serious bug corrected - maybe...
- As previously described a serious bug was detected causing seqtools to duplicate the sequence
part of a record overwriting the sequence of the following sequence in the project without
affecting its annotation. When the problem surfaced in March I rewrote the code performing
the navigation but apparently did not correct the problem. 21.08.2003 - Several minor improvements
- The batch sequence annotation facilities which auto-annotate sequences based on unattended
blast searches and retrieval and parsing of genbank sequence records are some of the very strong
features of seqtools. Some users have have asked for options enabling you to manually edit the
auto-generated annotation (genbank definition lines). This feature has been added to this
compile of seqtools. 07.08.2003 - Find antisense sequences in project
- A new function has been to the collection of search functions. The new function help you find
alignments after a blastn search where the project sequence is complementary to a genbank sequence.
This is critical when you plan to use project sequences for microarray oligonucleotide design. This
is primarily relevant when EST sequences are used for primer design. 24.06.2003 - Sequence names - The options for changing/modifying sequence names have been reorganised and extended. It is now possible to perform changes to sequence names on selected sequences in the sequence list. A new option has been included enabling you to use accession numbers, long sequence names, gene or locus names. In cases where these names are not available the original sequence names are used. 07.06.2003 - Clustering
- As a result of extending the number of allowed characters in sequence names to 16 in seqtools, the
reports generated by the clustering function produced scrambled output - or no output at all. This has
been corrected in the current revision. The three clustering methods include clustering as created by
the NCBI clustering program, clustering based on blast score values and clustering based on shared
identical sequence regions of specified length and with specified number of mismatches. 03.06.2003 - Duplicate sequences
- New functions have been added to identify and close duplicate sequences in a project. The form
includes three different options for identifying redundant sequences: About emboss - The comprehensive collection of programs for sequence analysis, EMBOSS, has recently been compiled for win32. These command line programs can very easily be integrated into seqtools. In case you need functions included in the emboss package, please let me know. 21.05.2003 - Minor bug fixes
- Bug fixes in formatting/verifying filenames. Small changes in microtiter plate index file function.
Implementation of EMBOSS functions under way. Change in NCBI script for retrieval of GI numbers and
sequence records implemented. Hopefully, the batch retrieval of sequence records from Entrez now works again. About emboss - The comprehensive collection of programs for sequence analysis, EMBOSS, has recently been compiled for win32. These command line programs can very easily be integrated into seqtools. In case you need functions included in the emboss package, please let me know. 18.05.2003 - Blast description line formatting
- When working with different projects the criteria for formatting information included in sequence lists often
different. Before is was necessary to compose the criteria used for trimming/selecting each time a different
project was loaded. An option is added to this revision enabling you to save and load formatting schemes used in the
sequence list settings. 09.05.2003 - New function added
- It is sometimes convenient to be able to transfer sequence information (selected subsections of the sequence header)
from one project to another. A new function has been added in revision 8.0.749 which allows you to import selected
subsections from an existing project saved as a SEQtools formatted multi-sequence file into the headers of
sequence contained in a new project. 05.05.2003 - Minor adjustments - Various minor adjustments have been made in this revision. 30.04.2003 - Description line formatting options extended - The description line formatting / editing options for sequence listing has been modified to improve flexibility. This has made it necessary to change the format of the ini-file (more space was required to hold the larger number of items in the words lists). When you start seqtools after this upgrade you will be asked to create a new ini-file for your project. This also implies that you must re-enter your preferences, sorry. About emboss - The comprehensive collection of programs for sequence analysis, EMBOSS, has recently been compiled for win32. These command line programs can very easily be integrated into seqtools. In case you need functions included in the emboss package, please let me know. 25.04.2003 - Context sensitive help updated - A few minor revisions are included in this revision - and the context sensitive help has been updated to cover the latest additions to SEQtools (several microarray related functions). There are, however, still several older functions where the context sensitive help provided does not exactly reflect the content of forms. 20.04.2003 - SEQtools folder structure, removing sequences from project
- The sub-folder structure of program and DOS folders has been changed to make subdivision of various data
types more logical. 14.04.2003 - Bug in create local db's, updated FastA def. line editor
- A bug preventing the proper operation of the function for creating local databases has been fixed. 02.04.2003 - Retrieving data from Entrez
- The utilities for data retrieval from Entrez (sequence records, medline records and GI numbers) have
been rewritten using the new cgi scripts made available by NCBI. Like the blast function the Entrez
retrieval functions have been made more robust resuming the retrieval if the first attempt failed.
The error-recovery handles server problems (time-out, no-response) as well as program errors. 22.03.2003 - Header search, Filtering of sequence lists, FastA definition lines, Help file
- The function for searching sequence annotation has been improved: You can now use boolean operators
and have the matches coloured in the displayed header. As before the first step in searching the
annotation is to compose the displayed header by selecting subsections to be included. 20.03.2003 - Alignment with ClustalW
- A small modification somewhere else in the code unfortunately made it impossible to select files for
aligning with Clustal. This problem is corrected in this update. 17.03.2003 - Duplicate filenames, remove sequences from project
- Handling of duplicate filenames improved: all routines dealing with verification of file/sequence names
in projects have been rewritten and hopefully now works as intended. The function removing sequences from
a project has been rewritten to improve speed. The microtiterplate indexing function has been improved to
facilitate indexing multiple plates from sequences contained in a project. 06.03.2003 - Serious bug detected
- I recently detected while processing a large set of sequences to
be included in a microarray, that rapid navigating from a long (10-50,000 bp) sequence to a short
(500-1,000) EST sequence in rare cases resulted in the replacement of the short nucleotide sequence by
the long. The annotation remained "correct" but the associated sequence no longer corresponded
to the annotation. 25.02.2003 - Fixes minor bugs
- A property value incorrectly set in the code prevented selection of multi-lines in the new sequence
listing form. This problem is corrected now. 23.02.2003 - Auto-installation of auxiliary programs
- SEQtools uses several third-party programs, contained in auxiliary.exe, for various functions, the
most important ones being the blast programs from NCBI for sequence comparison and ClustalW written by
Toby Gibson, Des Higgins and Julie Thompson for sequence alignment. These programs are not included in the
core download but must be installed after setting up SEQtools. 21.02.2003 - Auto-notification when a new SEQtools update is available
- After several attempts to implement his facility the automatic notification routine informing users about
new updates now seems to work (at least when I test it...). 20.02.2003 - Auto-retrieval of sequences from Entrez
- As a result of my current work designing new microarrays a number of routines have been added to SEQtools.
The functions enables you to "convert" various types of accession number lists to SEQtools
projects by auto-retrieving the nucleotide sequences from Entrez. 19.02.2003 - Improved sequence listing
- The design of the sequence listing function in SEQtools has long been very unsatisfactory. This rather
critical part of the program has been improved by replacing the old listbox control with the more
flexible listview control. 15.02.2003 - Sequence listing form redesigned
- It is now possible to sort the sequence list with an individual column as sort key. 30.01.2003 - Blast at NCBI - Bug in the NCBI blast function corrected. 03.01.2003 - SAGE analysis - Loading and analysis of SAGE profiles optimised. 26.12.2002 - Bug corrections - Loading of multi-sequence files substantially improved. A few bugs in the two functions for editing file names corrected. Several bugs in multi-database local blast search and result display corrected. The collection of multi-sequence tools optimised (but not yet tested very carefully...). A bug in the function converting enzyme file from GCG format to SEQtools corrected. 19.12.2002 - Bug corrections - Several bugs have been corrected in the latest update (8.0.671): the clustering functions now also handle protein sequences, the re-naming functions now accept sequence names up to 16 characters long, plus a number of smaller improvements most of then related to the increase in maximum length of file names (from 8.3 to 12.3/16). Speed has been improved by code optimisation in most functions (at least according to my code profiler) which also informs me that seqtools source code contains some 120,000 lines of which 66,000 are executable. No wonder that I sometimes feel a bit tired... 07.12.2002 - SEQtools published... - I wish to thank the large number of users for their kind remarks and encouraging comments to seqtools. There are probably still a number of corrections and adjustments to be made before the software package is fully functional. I hope you will continue to report any misbehaviour of the program as well as send me your suggestions for new functions. Remember to check the homepage regularly for updates. 22.10.2002 - SEQtools published - Rewritten from scratch SEQtools now appears for the first time as version 8.0. |