3. SEQTOOLS FEATURES
This page contains a number of general topics which could not conveniently be included under any of menu item title captions. In many cases more detailed/supplementary descriptions are found in one or more of the following pages.
- 3.1 organisation of the user manual
- 3.2 the dos folder
- 3.3 command line options
- 3.4 data files (restriction enzymes, codon usage tables)
- 3.4.1 restriction enzyme data files
- 3.4.2 convert gcg data file to seqtools format
- 3.4.3 codon usage tables
- 3.5 the main seqtools editor
- 3.6 project types
- 3.6.1 nucleotide / trace file projects
- 3.6.2 protein projects
- 3.6.3 primer projects
- 3.6.4 conversion of projects
- 3.7 working with projects
- 3.8 about sequence names
- 3.9 setting user preferences
- 3.10 sequence annotation (user comments, blast data)
- 3.11 batch operations
- 3.12 file types (recognised and/or created by seqtools)
- 3.13 application files and folders (created and maintained by seqtools)
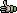 3.1 organisation of the user manual
3.1 organisation of the user manual
3.1.1 Introduction
This major revision of the seqtools manual comprise a complete reorganisation and rewriting of most topics of the manual including new screen shots of all seqtools forms. A long time has passed since the first version of the seqtools user manual was written. Since then a number of minor revisions have been made to the user manual in an attempt to cover new additions and modifications to the program. However, despite these efforts the application and its documentation now has diverged to an extent where major parts of the manual described features no longer relevant - and failed to mention important additions to seqtools.
As it is not nearly as interesting to write documentation as it is to build new facilities for the application this major revision has been postponed for a long time. The current manual was written February 2005 and hopefully will last for some time.
3.1.2 Organisation of the manual
Apart from the first three sections (1. Introduction, 2. Installation, 3. Features) and the last section (16. Primer) of the manual, the description of the various seqtools functions and facilities strictly follows the menu structure of the main editor form (section 3.5 below). This may not be the most optimal arrangement for the user, but hopefully makes it easier for me to keep the manual up-to-date in the future.
3.1.3 How to use the manual
Access to topics covered by the manual is by menu item caption of the main seqtools editor form. This retrieves in most cases a single page containing descriptions of all sub-topics included under the main topic. In some cases additional pages were necessary to cover special items which could not conveniently be contained on a single page.
The disadvantage of this organisation is that finding documentation to items not immediately identifiable by the menu or sub-menu caption is difficult. In such cases the context sensitive help may help guiding you towards the relevant section of the user manual.
3.1.4 User comments
In case you find that this manual insufficient you are welcome to contact me with criticism and preferably with constructive suggestions for improvements.
3.2 the dos folder
A number of SEQtools functions uses command line dos programs. To avoid problems with the length of file paths (many dos programs are unable to handle file paths unless they follow the old 8+3 syntax) all such programs and associated components reside in a special SEQtools folder on the c drive under the folder containing the operating system (WINNT, Windows): C:\WindowsFolder\ST8_TEMP.
When SEQtools starts it checks whether all necessary external components are available in this folder. If components are missing the user is warned and encouraged to download the missing components. The components are contained in two self extracting compressed files, auxiliary8.exe and emboss8.exe.
When you install the full SEQtools packages you automatically install also these components. New updates of the auxiliary and emboss programs and be downloaded and installed without re-installing SEQtools. Use the functions under the Help/SEQtools Configuration menu to perform this task.
3.2.1 Moving the dos folder to a new location
It is possible - but not recommended - to move the SEQtools dos folder to a
different location. If you prefer the dos folder to be located in a different location
use the Preferences/General Preferences/DOS Directory to choose a new location.
Click Accept to copy the entire content of the ST8_TEMP folder to the new location.
Note that the new path must follow the standard dos syntax (8+3) to pass the verification
routine before the new path is accepted.

3.2.2 Components located in the dos folder
The following sub-folders and files must present in the SEQtools dos folder:
| \ST8_TEMP\data\*.* | Contains the 26 NCBI data files required by the different NCBI programs |
| \ST8_TEMP\DB\*.* | Contains local databases created by formatdb. Each local database consist of 5 files all with the sane name but with different extensions |
| \ST8_TEMP\EMBOSS\... | Includes two sub-folders: acd containing four acd files and data containing five data files required by emboss programs |
| \ST8_TEMP\TMP\*.* | Contains temporary files created by different SEQtools functions. The TMP folder is cleared when SEQtools closes |
| \ST8_TEMP\*.* | Contains executables and dll's for a number of components used by SEQtools |
3.3 command line options
SEQtools creates and saves a specific ini-file for each instance of the program.
This implies that you can create pre-defined instances of the program for different sequence
types. Note that you must create a new icon on your desk
top with the instance parameter (/I=NN) before you
open the SEQtools instance to set the preferences for the instance.
Proceed as follows: Create a new SEQtools icon on your desktop. Right-click the icon to
display the Windows pop-up menu. Left-click the Properties line of the pop-up
menu and edit the load path for the program as described below. Then launch the
SEQtools instance, set the preferences and exit SEQtools to save the ini-file
associated with the new instance.
valid command line parameters:
1. SEQtools instance number (/I= (00 - 99)
2. full path to sequence file to load when SEQtools opens
examples:
set project type
c:\app.folder\seqtools83.exe /I=00 (main instance, default)
c:\app.folder\seqtools83.exe /I=01 (nucleotide project)
c:\app.folder\seqtools83.exe /I=02 (protein project)
c:\app.folder\seqtools83.exe /I=03 (primer project)
load specified file
c:\app.folder\seqtools83.exe c:\mydir\myfolder\my_sequence.seq /I=05
c:\app.folder\seqtools83.exe c:\mydir\myfolder\my_project.fms /I=10
c:\app.folder\seqtools83.exe c:\mydir\myfolder\my_protein.seq /I=15
c:\app.folder\seqtools83.exe c:\mydir\myfolder\my_primer.seq /I=20
c:\app.folder\seqtools83.exe c:\mydir\myfolder\my_project.plp /I=25
3.4 data files
Seqtools uses two types of data files: restriction enzyme data files and codon usage table files. When SEQtools is installed four restriction enzyme files and four codon usage files are included in the installation. The Data files are located in the main application folder in the ...\Program Files\seqtools 8.3\DataFiles\EnzymeFiles\ and the ...\Program Files\seqtools 8.3\DataFiles\CodonFiles\ sub-folders. Seqtools uses its own file format and both file types must thus be processed before they can be used in the program as described below.
3.4.1 Restriction enzyme data files
Updated restriction enzyme data files can be downloaded
fromReBase.  In addition to enzyme data files, the ReBase homepage contains a very useful search function which allows you to search their data base with the name of an enzyme or with a recognition pattern. Visit theReBasehomepage to download the restriction enzyme data file in GCG format.
In addition to enzyme data files, the ReBase homepage contains a very useful search function which allows you to search their data base with the name of an enzyme or with a recognition pattern. Visit theReBasehomepage to download the restriction enzyme data file in GCG format.
3.4.2 Convert gcg data files to seqtools format?
Seqtools uses a slightly different enzyme data file format than the GCG program so it is necessary to use Tools/Conversion Functions/Convert GCG Restriction Enzyme File... to convert the file format so that the data file can be used by SEQtools as illustrated by the three screen shots below:
3.4.3 Codon usage tabels
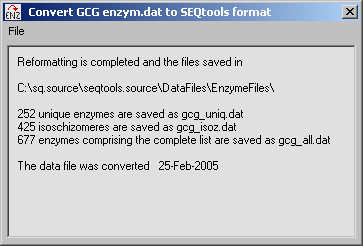
Codon usage tables can be obtained from a number of sources for example from the Japanese Kazusa DNA Research Institute/Codon Usage Database. Remember to specify a GCG like style. The easiest way is to save the table directly from the Internet browser window as a plain text file with the extension *.cod in the folder ...\Program Files\seqtools 8.3\DataFiles\CodonFiles\mycodons.cod. Note that some browsers adds a *.txt extension to the file in addition to the *.cod extension you typed (...myfile.cod.txt). To avoid this enclose the filename+extension in quotes before saving from the browser.
3.5 the main seqtools editor
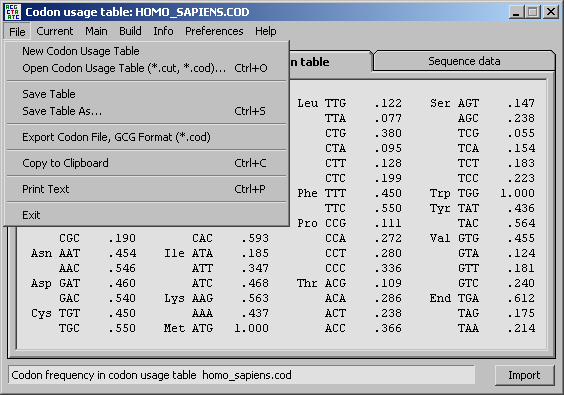
Below is a screen shot of the main SEQtools editor. The form includes of a sequence panel, a sequence list (right clicking the sequence list toggles between a sorted list, a project order list. Pressing <F5> lists the matches from a local blast search). In the lower part are two info fields, a goto/bookmark field, an editable sequence name field and command buttons for an extended sequence list, the sequence header, chromatogram display and navigation buttons. The Update button reformats the sequence after editing.
Parking the cursor over the upper info field and holding down the right mouse button retrieves blast information for the displayed sequence (if the information is available). The vertical panel to the right contains shortcuts to a number of commonly used functions.
3.6 SEQTOOLS project types
Before you create a new SEQtools project you need to decide which type of sequences you wish the project to contain. In cases where you load a project which is previously generated and saved from SEQtools, the SEQtools auto-detects the project type from the first sequences in the selection and sets editor options accordingly.
It is not possible to mix nucleotide, protein or primer sequences in the same project. If you which to work with different sequence types simultaneously, open separate instances of SEQtools - one for each sequence type and use copy/paste to transfer sequences of the same type between the separate instances of SEQtools.
3.6.1 Nucleotide / trace projects
This project is restricted to include nucleotide sequences. If protein sequences are generated by translation of nucleotide sequences the protein sequences do not become part of the project when the project is saved.
Extracted trace files (chromatograms from auto sequencers) require a nucleotide project. If you create a new project exclusively consisting of trace files SEQtools auto-detects the project type and create a trace project. A trace project is similar to a normal nucleotide project. You can add more normal sequences and new trace files to a trace project and save the entire mixed project by one of the four methods described below.
The original trace file is not modified by being loaded and saved from SEQtools. Instead an association/link is created between the extracted, normal SEQtools version of the trace sequence and the original trace file. Provided that the path to original trace file is not changed the chromatogram can be retrieved and displayed by clicking the trace icon on the main editor form.
If you attempt to load a non-nucleotide sequence into a nucleotide project you are warned before SEQtools cancels the load operation.
3.6.2 Protein projects
This project type is limited to protein sequences. If you attempt to load a non-protein sequence into a protein project you are warned before SEQtools cancels the load operation.
The project type is auto-detected by SEQtools based on the first sequence in the load selection or a multi-sequence file.
3.6.3 Primer projects
This project type only holds primer sequences. If you attempt to load a non-primer sequence into a primer project you are warned before SEQtools cancels the load operation.
3.6.4 Conversion of projects
It is possible to convert primer projects to nucleotide projects and vice versa. This option is useful if you for example want to perform a blast search at Genbank with a collection of primer sequences.
Note, however, that due to the different structure of sequence and primer headers converting a nucleotide project to a primer project - and saving the project as a primer project will lead to irreversible loss of all information contained in the original sequence headers.
The fact that several symbols (brackets, IUB symbols) which are allowed for primer sequences but not in normal nucleotide sequences implies that the conversion option should be used with caution, especially when converting normal sequences to primers and primers with degenerate positions to sequences.
3.7 Working with projects
3.7.1 Create projects
A SEQtools project is automatically created when you load a collection of sequences into the program. This can either be done by navigating to a specific folder and selecting one or more sequence file, by loading a multi-sequence file or by creating an empty sequence file and entering the sequence by manually typing or by copy/paste.
3.7.2 Modify projects
It is possible to add more sequences to an existing project by importing multiple single sequences, multi-sequence files or by creating new sequences manually. New sequences added to a project are appended to the sequences already included in the project.
Sequences can also be removed from the project by selecting the sequences to be removed from the project in the sequence list and click the remove icon. Note that removed sequences remain on the hard drive, i.e., are NOT deleted but only excluded from the project.
With this facility it is for example possible to perform a database search with all sequences contained in a given project - and remove sequences with matches worse than a specified expect value.
3.7.3 Save/export projects
Sequences can be saved/exported in three different ways:
- - as single sequences,
- - as a multi-sequence file in fasta or SEQtools format
- - as a so called pfp file which is a list containg the full path from which the sequences in the project were imported or
- - a psp file which also consist of a path list, but in this case the save-path for all project files.
The latter option is not enabled until the project is saved as single files.
Note for the pfp and psp save methods that changing the physical location of the sequence files on the hard disk after the pfp and psp file are generated will prevent these sequence files from being loaded from the pfp and psp path-list file.
3.8 About sequence names
3.8.1 Normal sequence name - Most of the functions related to handling multiple sequences in SEQtools were developed during a small EST project
carried out at the Carlsberg Laboratory. The purpose of the project was aimed at obtaining information about the Blumeria (mildew)
genome and gene expression to better understand the interaction between the obligate plant parasite and its host, barley.
All the clones from the cDNA libraries used in the analysis were sequenced twice, with an F (forward) primer and with a R (reverse)
primer. The 5' sequences of the insert were used for database searching for homology in public data bases while the 3' (polyA)
sequences were used to create links to SAGE profiles generated from the same developmental stages.
The insert lengths of the cDNA libraries were rather short (only very few were full length ORFs) which turned out to be an
advantage when searching the international databases. It also implied that in many cases the F and R sequence overlapped
and could be replaced by the merged, complete sequence of a particular insert. This feature of the libraries allowed us to
replace the F and R sequences by their merged sequence which both improved the quality of the sequence and reduced the number
of sequences in the Blumeria database.
In cases where the F and R sequence of a clone/insert did not overlap, i.e., where sequence information was not available to
link F and R sequences from the same clone, the file name was used instead as a link between corresponding F and R sequences
of the same insert. Obviously this requires that files / clones must be named consistently as described below.
In order to keep track of the F and R sequences originating from the same insert/clone, all sequences were named using
-F, -R and -M to indicate the 5', 3' and merged sequence.
3.8.2 Long sequence name - Loading a new sequence with a long, non-DOS, file name into SEQtools automatically transfers the
long file name into the Long name variable of SEQtools.
For new sequences which have not previously been formatted by SEQtools, a Long name is automatically created consisting of the
file name followed by the number symbol (#) and a random 8-digit number (e.g. C00018-F #47382957). The Long Sequence name cannot
be changes by the user.
3.9 Setting user preferences
There is a number of options for the user to customise the appearance and behaviour of SEQtools through extensive
preference facilities.
These options are described in details under the Preference menu item. At this point it suffice to briefly mention which aspects of SEQtools behaviour that are adjustable by preference settings.
| General settings | Project files, Colors and fonts, Launch applications, Launch URLs, Backup settings, Checksum calculation, DOS folder location, Footnotes. |
| Project settings | Trace file folder, Global timeout, Project blast settings, Project title, User data, Sequence format, Color patterns, Header |
| Form behaviour settings | Main editor, Header forms, Blast forms, Compare forms, Tools, Translate, Primer forms, Special function forms |
| Description line format | General settings, Left-trim lines, Right-trim lines, Replace lines |
| Chromatogram import settings | Basecallers, Preset options, Trimming, N-threshold, Gap-quality |
| NCBI inifile settings and editor | Inifile settings for blast searches |
| Internet connection and servers | NCBI settings, Internet connection test URL |
| Compose search data file | Predefined groups, User defined groups |
| Log and Ini-file viewer: | Ini-files for multiple instances of SEQtools, Log-file for several batch functions |
| Application color coding | Assign color schemes to multiple instances of SEQtools |
3.10 sequence annotation
3.10.1 Auto-annotation
Seqtools includes various methods of auto-annotating sequences. The most powerful ones are the batch blast functions which allows you to perform unattended blast searches at NCBI/Genbank with a large number of sequences. Depending of your pc you can load 10 - 20.000 sequences into a single project and perform batch blast search on them all. Seqtools stores this information in the sequence header associated with each sequence.
Seqtools contains advanced facilities for handling and displaying this information. It is possible to select a particular blast search and list or display this information excluding/hiding results from other search results stored in the sequence header. As all information is stored in the RAM memory of your pc processing large amount of sequences requires quite a lot of RAM.
Information stored in sequence headers can be search in a number of ways making this a very flexible system. You can read more about this in the special Header section of this manual
3.10.2 User annotation
It is possible to manually enter your own comments and copy/paste external information into sequence headers. This, however, must be done sequence-by-sequence.
3.11 batch operations
One of the strong features of SEQtools is the facilities for performing batch operations. A number of
tasks such as changing sequence names and performing blast searches locally or on databases at Genbank can be
performed without user intervention. Some users have reported successful batch analysis of as many as 30,000 est
sequences in a single job running over several days.
The fact that you can launch several concurrent instances of seqtools makes it possible for example to run large
blast search jobs at genbank while performing other analyses with a different instance of seqtools.
You can even run parallel batch search jobs at genbank with separate instances of seqtools. Seqtools only uses
very few pc resources for processing and storing search results as they arrive from ncbi. The auto-save function
of the batch blast function reduces the risk of loosing data in case of pc craches during a search job.
3.12 seqtools file types
Seqtools uses a number of different file types, some of them for saving various types of data others for importing data. The table below lists the extension of file types recognised or created by SEQtools.
| ALN, PIR, PHY, MSF | Output files from sequence alignment with Clustalw. |
| DAT, SDF, GCG | Restriction enzyme and user created search data files. |
| PLP, PSP | Project path files, used to store the full paths for all files in a project for reloading the complete project or a sub-group of the project. |
| FOF | File of files. Includes a list including the names of all sequences included in the project. |
| TXT, RTF, LST, RPT, LOG, TAB | Various ASCII files containing sequence lists, reports, logs etc. |
| SEQ, DNA, PRO | General extensions for DNA or protein sequence files. |
| CUT, COD | Codon usage tables, SEQtools and GCG format. |
| FMS, FMZ, TMS, MSF, DMS, FAS, FSA, GB, LGF, GBK, GCG, MBL, FMS | Various types of multi-sequence files. |
| B!!, BA!, BAK | Backup files from timed project auto-backups. |
| TPL, ESF | Template and complete submission file for transfer of EST sequences to Genbank. |
| STF, PTF, DTF, MTF, SMF, CGI, TDT | Extensions used in SAGE related functions. |
| SGD, MCA | Extensions used for files created by EST clustering functions. |
| OOF, COF, MSG | Primer mail order files. |
| BMP, WMF | Image files. |
| MTP, MPF, IGF, IMG | Microtiter plate index, Microarray project file, Imagene GeneID file |
3.13 Application files and folders created and managed by seqtools
| \windows\NCBI.ini | Ini-file for blast programs. |
| \app\ST8##.INI | Ini-file for instance ## of SEQtools. Contains all user preferences for instance ##. Each instance (maximum number of open instances of SEQtools is 99) has its own set of preferences. |
| \app\ST8_instances.dat | Seqtools session dat-file. Keeps track of open instances of SEQtools. |
| \app\BackupData\ | Contains timed backup files for open / active projects. Each instance of SEQtools has its own timed backup file. |
| \app\DataFiles\CodonFiles\*.* | Codon usage tables. |
| \app\DataFiles\EnzymeFiles\*.* | Contains all restriction enzyme data files. |
| \app\DataFiles\genbank_databases.dat | Contains a list of available Genbank databases for advanced batch database searching at Genbank. |
| \app\UserData\NNN\*.* | Auto-generated default folders for storing various data: _array _blast _cluster _database _default _genbank _multiseq _primer _protein _psgfiles _sage |
© 2002-2010S.W. Rasmussen (revised: )