4. SEQTOOLS MENUS
This page contains a brief summary of the functions grouped under each of the 15 menu titles for nucleotide projects. The menu layout is slightly different for protein projects and more so for primer projects. Some menu items are not displayed when seqtools is opened for handling protein and primer sequences. The manual contains a separate page, primer functions, describing specific facilities related to primer design and ordering. The differences in menu layout are smaller for protein projects and are not treated separately.
- 4.1 file menu
- 4.2 edit menu
- 4.3 translate menu
- 4.4 search menu
- 4.5 retrieve menu
- 4.6 compare menu
- 4.7 analyse menu
- 4.8 header menu
- 4.9 project menu
- 4.10 launch menu
- 4.11 tools menu
- 4.12 special menu
- 4.13 www menu
- 4.14 preferences menu
- 4.15 help menu
 4.1 file menu
4.1 file menu
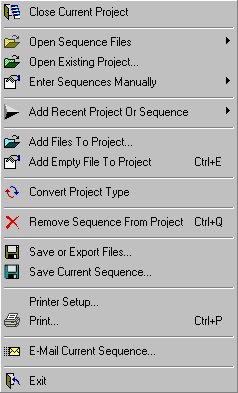
The File menu contains a number of facilities for creating and modifying projects. With these functions you can select sequences to be included in a project, add more sequences to an existing project or remove selected sequences from a project.
Sequences can either be loaded as collections of single sequence files, as multi-sequence files or a mixture of both. Seqtools examines each file to be loaded to see if it contains a single sequence or is a multi-sequence file.
If you need to enter sequences manually or by copy/paste it is necessary first to create an empty file to hold the sequence.
Multi-sequence files can either contain the complete sequence and annotation for each sequence in the multi-sequence file or be a list (a plp-file / psp-file) of file-paths to each sequence file.
In the first case all project files must be located in the same folder while in case of plp-files / psp-files the file paths can point to sequence files located in different folders.
The save and export facilities allow you to save / export sequence files in the most common sequence formats.
4.2 edit menu
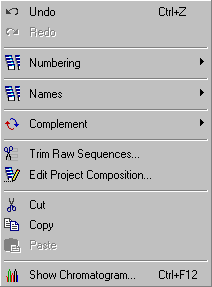
The Edit menu includes functions related to sequence editing in the broadest sense.
With these functions you can edit sequence names and numbering, remove vector parts of raw sequences generated by auto-sequencers (Trim Raw Sequences...), convert one or more sequences to their complementary sequence and remove sequences from the project based on sequence quality (Edit Project Composition...).
Most of the functions for sequence editing apply to nucleotide sequences and are not visible when seqtools is in protein mode.
4.3 translate menu
![]()
The Translate menu contains a number of options for translating nucleotide sequences into protein. In addition it allows you to rapidly find the longest open reading frame or the longest stretch without stop codons in an unknown nucleotide sequence.
If a protein sequence is displayed you can back-translate it into a nucleotide sequence if you provide information about the expected codon usage in the form of a codon usage table either retrieved from a web resource or created by yourself.
As for the Edit menu above, most of the Translate functions apply to nucleotide sequences and are not visible when seqtools is in protein mode.
4.4 search menu
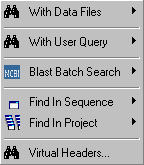
The Search menu includes a number of advanced options for searching with your nucleotide or protein sequences.
The functions range from trivial searching your sequence with a query string to unattended batch blast searching all sequences in the project against Genbank or against a local sequence database created by yourself.
In addition you may look for repeats, introns and similar/identical sequences in the project.
Some of these functions uses programs included in the Emboss collection others depend on the NCBI program collection.
Batch searching Genbank requires an Internet connection.
4.5 retrieve menu
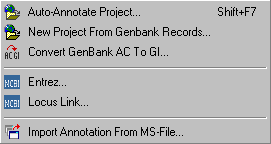
The Retrieve menu includes various functions for annotating your sequences and for auto-editing already existing annotation.
With these functions you can retrieve the complete annotation from Genbank if your sequences are only identified by their Genbank accession number.
Or you can automatically create a new project consisting of the Genbank sequences with the best match from a blast search on Genbank with your own sequences.
4.6 compare menu
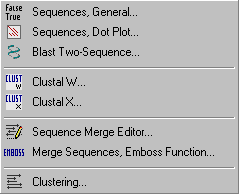
The Compare menu comprises several functions for comparison and/or alignment of two or more sequences.
The functions include two-sequence comparison, multi-sequence alignment with ClustalW or ClustalX, the former option with optional post-processing of the alignment with t-coffee.
The Compare menu furthermore contains two functions for multi-sequence merging, one based on the Emboss program the other a simple multi-sequence editor.
Finally, this menu includes functions for sequence clustering based on different methods.
4.7 analyse menu
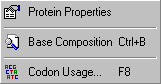
The Analyse menu lists three functions which enables you to view basic properties of a displayed protein sequence, the base composition of a nucleotide sequence and the codon usage of a nucleotide sequence.
The latter function furthermore enables you to create a new codon usage table and to include the codon usage of the currently displayed nucleotide sequence in this - or an already existing - codon usage table.
4.8 header menu
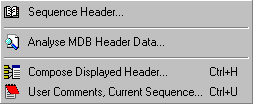
Different options related to handling sequence annotation is collected under the Header menu. This includes display of the items currently selected as the virtual header, options for displaying the result of local multi-database searches and a form enabling you to enter your personal comments to the sequence.
Finally, the "Compose Displayed Header..." option allows you to select items of the complete annotation to be included in the virtual header.
4.9 project menu
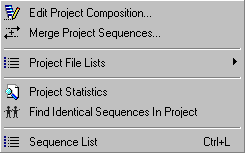
The Project menu includes functions and options related to the handling of the sequences contained in the current project.
With these functions you can create lists of sequence names and file paths, merge overlapping forward and reverse sequences from the same insert, calculate project statistics, find (and remove) duplicate sequences in the project (irrespectively of the sequence names) and display a list of the sequences contained in the project.
The latter function is quite elaborate enabling you to include selected information from the current virtual with extensive options for formatting the displayed sequence list.
4.10 launch menu
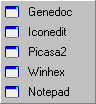
The Launch menu contains up-to five third party programs selected from the "General Preferences...".
Two external programs (GeneDoc and TreeView) are recognised by seqtools and can be accessed from the ClustalW alignment result form. T-coffee mentioned above under the Compare menu is also accessed from the ClustalW result form but do not have a user interface. This program is included in the seqtools installation and need not be installed separately via "General Preferences...".
4.11 tools menu
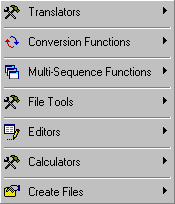
Various functions and facilities have been collected under the Tools menu. These include a codon-amino acid translator, a IUB symbol translator and three converters (GCG restriction enzyme file -> seqtools format, project -> search database, Genbank accession numbers -> GI numbers).
The menu also includes several tools for multi-sequence handling (building of local databases, batch-editing of sequence annotation, building new projects from Genbank records). Among the "File Tools" are various facilities for viewing, searching and creating different file types.
The "Editors" include options to customise restriction enzyme search datafiles and to compose/edit FastA definition lines for multi-sequence files.
Finally, the "Create Files" item covers functions for submission of EST sequences to Genbank and a multi-sequence annotation parser.
4.12 special menu
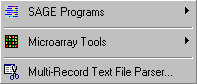
The Special menu contains a number of functions and facilities for handling and processing data for SAGE (serial analysis of gene expression) and for oligo-nucleotide based micro-array analysis of gene expression.
4.13 www menu

The WWW menu is a list of URL to selected resources on the Internet.
You can customise the list from the "General Preferences..." form. The list holds up-to ten URL's.
Genbank resources are indicated by NCBI-icons and others by globe-icons.
4.14 preferences menu
The Preferences menu includes a series of forms containing options for customising the behaviour and appearance of seqtools.
Most of the menu items are self-explanatory, other more obscure. Among the latter category is the "Form Behaviour Settings..." which enables you to decide if a given seqtools form should always stay on-top of other form on your desktop.
In some cases the "NCBI Settings, Firewall..." are important to establish an Internet connection through a local firewall.
The "Application Color Coding" allow you to color code different instances (one instance in primer mode, a second in DNA mode and perhaps a third instance running in protein mode) of seqtools running simultaneously on your pc to facilitate identifying each instance of seqtools.
It is highly advisable to invest in a second monitor if you are using seqtools regularly, especially when you run several instances of the program simultaneously.
4.15 help menu
The Help menu contains both different help items, options for registering seqtools (entering the registration key in the program as well as on-line renewing your registration) and a form listing the current seqtools configuration (file dates and installed auxiliary components) and program update options.
You also find a form for reporting bugs on the help menu.
The "Animated Demos, Viewlets..." menu item only contains two animated sequences illustrating program basics. It has been my intension for a long time to write more viewlets describing other aspects of seqtools. It is, however, quite time consuming to produce viewlets so you may have to keep waiting for more animations.
© 2002-2010S.W. Rasmussen (revised: )