4.1 FILE MENU
- 4.1.1 about files and projects (general comments)
- 4.1.2 close current project (without exiting SEQtools)
- 4.1.3 open sequence files (to create a new project)
- 4.1.4 basecalling chromatograms (processing trace files)
- 4.1.5 open existing project (from list of file paths)
- 4.1.6 enter sequences manually
- 4.1.7 load/add recent project or sequence (selected from list of recently opened files)
- 4.1.8 add more files to a project (using the file selection form)
- 4.1.9 add an empty file to a project
- 4.1.10 convert project type (primer to dna / dna to primer)
- 4.1.11 remove sequence from project
- 4.1.12 save project / export files
- 4.1.13 print project
- 4.1.14 e-mail current sequence
- 4.1.15 close project and exit
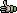 4.1.1 about files and projects
4.1.1 about files and projects
A project in SEQtools is simply a collection of one or more sequences of the
same type (nucleotide, protein or primer). It is not possible
to include different sequence types in the same project. If you wish to create a project
from more that one sequence file, all files to be loaded must be located in the same
folder.
You can add more sequence files to an existing project from other
folders. In most cases SEQtools will auto-detect both the file type (nucleotide, protein
or primer), sequence format (SEQtools, embl, fasta, genbank, etc.) and file format (single,
trace, multi-sequence - or a mixture of the three) and create the project from the selected
files without your intervention.
Saving a project is most conveniently done by using the standard SEQtools multi-sequence format
which saves all sequences in the project in a single file (with or without compressing the
file).
The file menu contains the following menu items (described in more detail in
separate sections below):
4.1.2 close current project
SEQtools issues a warning before closing the current project offering to save the sequences. Closing a project without saving the data will cause irreversible loss of editorial changes to the sequences as well as all information added to the sequence headers.
4.1.3 open sequence files
Sequence files to be included in a project can be selected in different ways as
indicated in the screenshot of the Open Sequence Files menu shown below.
SEQtools attempts to determine sequence type and format and file format before loading the data into a new project. In most cases this does not require user intervention provided all sequences to be loaded are of the same type (nucleotide, primer or protein).
The project type (nucleotide, primer or protein) is determined by the first sequence loaded. If a sequence of a different type is encountered a warning is issued and loading is interrupted.
SEQtools recognises and loads four sequence formats either as single sequence files or as collections of sequences in multi-sequence files: SEQtools, EMBL, Genbank and Fasta
Before the file selection form is loaded the Project Preferences form
is opened to enable you to give the project a title and to set various parameters for the new
project.

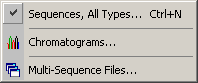
The File Selection form is used to select the sequence files for the
project. A drive list box and a file list box allows you to navigate between
drives and directories to locate the sequence files you wish to include in the project.
The top file list contains all files in the selected directory. The bottom file list shows
the files currently selected for loading.
Files are selected from the directory file list by pointing or dragging the mouse pointer to highlight one or more file names. A discontinuous series of files is created by holding down the <CTRL> key while clicking the filenames to be included in the project. Clicking the Add Files command button activates the selection. File names can be removed from the list of selected file names by clicking the file name.
Files with the following extensions (cab, log, fof, exe, ini, sys, com, hlp, bat, oof, cof, msg, cut, cod, lst, zip, dat, qscore.fasta, gap_qscore.fasta) cannot be selected and loaded into a project unless the Options/File Exclusion Enabled/Disabled option is set to File Exclusion Disabled.
It is possible to add a case-insensitive filter to the selection by typing characters in the text field. Only files which include or do not include - depending on the selected option - these characters in their file names will be selected/deselected when the Add To List command button is clicked.
When the auto-backup option is active (Preferences/Project
Settings/Timed Backup) a complete backup of all sequences and sequence headers of
the project is saved - at the specified time interval - to a multi-sequence file (*.fms)
located in the main application folder (normally c:\SEQtools 8.3\BackupData\).
If you need to load a backup copy of a previous project select the Load project
backup file(s) option on the load form to set the path to this folder and load the *.fms
multi-sequence file into a new project.
If you are loading more than 300 sequences into a project, SEQtools offers to turn off the
timed backup function. This function is often not required for large projects and turning it
off saves resources for processing other functions.
When selection is completed, clicking the Load Files command button causes the selected files to be loaded into the specified project. It is not possible to select the same file twice nor is it possible to select files from different directories when a new project is created. Additional files can be added to the project later.
If you already know that the sequences to be loaded are contained in a multi-sequence file (SEQtools, Genbank or Fasta format) just select the Multi-Sequence Files... menu item. This opens a standard Windows file dialog box for selecting the multi-sequence file. The file selection form is not loaded in this case.
It is possible to select and load a mixture of normal single files and multi-sequence files.
When sequence loading is completed and a new project created SEQtools
displays a summary of the annotation (primarily a list of blast search results) available
for the loaded sequences. This is described in more detail under 4.8 Header menu
and its sub-items.
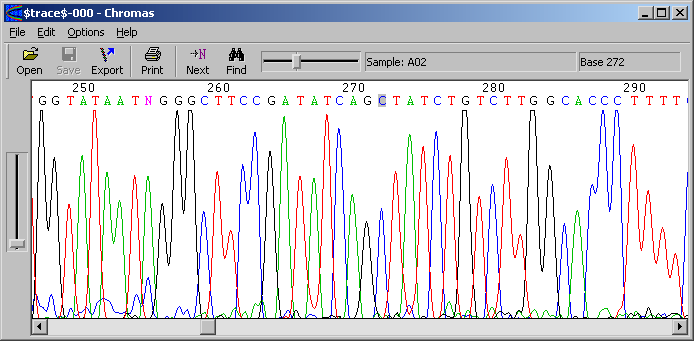
4.1.4 basecalling chromatograms
SEQtools auto-detects if the file to be loaded is a chromatogram produced by an
automated sequencer. Extraction of the plain DNA sequence from the trace file is, by default,
carried out by the convert_trace program from the Staden package while viewing the
traces is done by Chromas (see screenshot below).
The link between the extracted sequence and the chromatogram is the Long
Filename of the sequence and the path to the trace file folder set in
Preferences/Project Settings/Trace File Folder.
Provided this association is intact the chromatogram can be retrieved later
and viewed with the Chromas program.
To maintain this connection it is important that the long sequence name is not changed in SEQtools. If you alter the long file name for a sequence, the link is broken and can only be re-established if you enter the name of the trace file corresponding to the SEQtools sequence again.
If you want to check a certain position in your sequence against the
chromatogram, highlight the region in the main SEQtools editor and press CTRL+C
to copy the region to the clipboard. The highlighted region in the sequence is
coloured blue to facilitate locating it.
In Chromas, click Edit/Find... to display the search form. Press CTRL+V to
paste the selected region of your sequence into the search form of Chromas and
click Find. SEQtools removes spaces, CR, LF, and numbers from the selected region,
so it does not matter if your selection spans two lines.
The advantage of keeping SEQtools formatted sequences and the original trace files separate is that all SEQtools functions, including automated annotation for example generated by blast searching can be maintained in the sequence headers.
4.1.4.1 convert_trace
4.1.4.1 Convert_Trace is the default program used by SEQtools to extract plain nucleotide information from chromatogram files. The extracted nucleotide sequence is generated by the basecalling performed by the application which created the chromatogram and does not allow the user to modify/adjust the way the basecalling is carried out.
4.1.4.2 lifetrace
4.1.4.2 LifeTrace on the other hand is a stand-alone basecaller which uses information
included in the chromatogram to perform de-novo basecalling utilising its own algorithm for
calling bases.
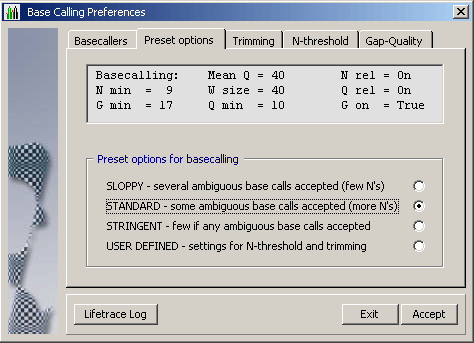
LifeTrace runs on Linux/Unix systems and requires a more complex setup than convert_trace. In brief: Sequences must be copied to a Linux/Unis computer running LifeTrace to generate the data files used by SEQtools to post-process the basecalling. The advantage is that the user has full control over the basecalling operation as well as of the post-processing by SEQtools. Take a look at the preferences form above to get an impression of the options available when LifeTrace is used for basecalling/extraction of the nucleotide sequence from a chromatogram.
LifeTrace is particularly effective when applied to MegaBACE capillary sequencing machines. A detailled description of the LifeTrace /SEQtoolssetup and interactionand thecommandline argumentsare given on separate pages of this manual.
4.1.5 open existing project
If a *.psp (project save paths) or a *.plp (project load paths) for
a project exists it is possible to re-open the project from the Open Existing Project
menu. The *.psp and *.plp files are lists of full paths to all sequence files included in
the project. The files may be located in different directories and can be single or
multi-sequence files - or a mixture of the two types.
The *.plp and *.psp files can be saved by clicking the Project/Project File Lists
as shown by the screenshot below.
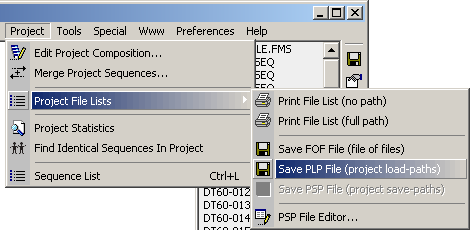
The *.plp file is auto-generated when the project is created while the *.psp
file is auto-built/re-built each time the project is saved. This option is enabled in
Preferences/General Settings/Project Files

4.1.6 enter sequences manually
In case you wish to enter sequences manually either by typing the sequence
or by copy/paste from other applications or from additional instances of SEQtools you need
to tell SEQtools which type (nucleotide, primer or protein) of sequences you intend
to include in the project. When you choose this option, SEQtools sets the project type and
opens an empty file ready for receiving the new sequence.
Each additional sequence requires that you first create a new, empty, page (see below) to
hold the sequence before you start typing or copy/paste. Remember that a project can only
hold one type of sequence
4.1.7 load recent project or sequence
SEQtools stores the last 20 opened sequence files (single and multi-sequence)
in the Open Recent Project or Sequence list for easy loading of often accessed
files. It is only possible to select and load one file from the list at a time. Note
that this list may include sequence files belonging to different sequence types.
The different sequence file formats are indicated by different icons. To clear the list
of recently opened files, click the title line of the list.

4.1.8 add more files to a project
Once a project is created more sequence files can be added to the project
using the load form described in sections 4.1.3.. and 4.1.5. Note, however, that using
the 4.1.3 sub-menu will close the current project and create a new SEQtools project
while the Add Files To Project... add the selected files to the existing
project.
Apart from this difference the load form works exactly in the way described in
section 4.1.3.
It is also possible to add more sequences to the project using the Add Recent
Project Or Sequence
While adding sequence files to the project SEQtools warns you if you load
sequences with filenames already present in the project. If you choose to override
the warning and accept multiple files with identical names, SEQtools will modify
the filenames of such files if the project is saved as single sequence files in order
to avoid overwriting the first saved file with subsequent sequence files with the
same name.
Notice that the file type (nucleotide, primer or protein) of files to be
added to an existing project must be of the same type as the files in the project.
Sequences loaded with this function are appended to the list of sequences already
in the project.
4.1.9 add an empty file to a project
Before you can add sequences to an existing project by typing the sequence or by copy/pasting the sequence from a different source you must first add an empty page to the the project to hold the sequence. Click Add Empty File To Project to append an empty page to the end of an existing project.
4.1.10 convert project type
Occasionally it is convenient to be able to perform a blast search on Genbank
databases with oligonucleotides designed for microarrays. This can most easily be done by loading
the oligonucleotides into a primer project in SEQtools and subsequently convert the project to
a nucleotide project. This function Convert Project Type enables you to convert primer
projects to nucleotide projects and vice versa.

Important note: Converting a nucleotide project to a primer project will irreversibly remove all information stored in sequence headers due to the different design of the header structure of the two project types in SEQtools.
4.1.11 remove sequence from project
To remove a single sequences from a project simply highlight the sequence to be removed in the sequence list and click Remove Sequence From Project. The removed sequence is not removed from the hard disk, just no longer a member of the project.
To remove a selection of sequences from a project proceed as follows:
Hold down <CTRL> while clicking the sequences to be removed. <Shift+Right-Click> on
the sequence list to open the pop-up menu. Select Close Selected Sequences to remove
the selected sequences from the project. Again, the sequences are not deleted from the hard disk but only
removed from the project.

4.1.12 save project / export files
This function File/Export Formats formats the sequence and its header
so that they can be loaded into other nucleotide and protein analysis programs. There is a special
function which allow you to customise the single line header - the Definition Line - used
in Fasta format.
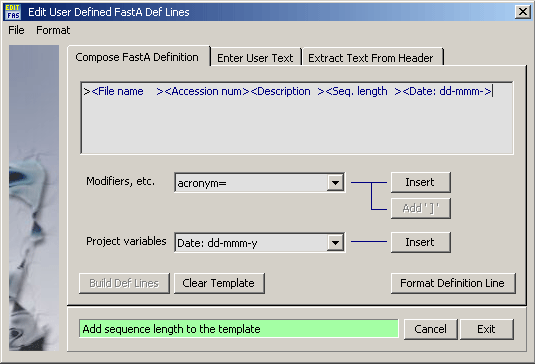
The different save/export formats supported by SEQtools are shown in the
screenshot of the save/export form. Additional options are available for several of the
export formats. Among these is an option for compressing multi-sequence SEQtools files which facilitates loading the file into a SEQtools project and
saves disk space.
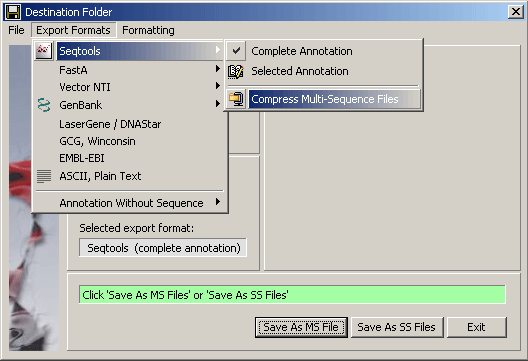
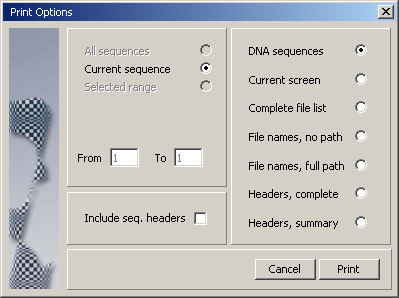
4.1.13 print project
Printing projects is usually not a relevant option. In most cases the amount of
data included in a project makes printing meaningless. As a consequence the printing facilities
in have not been revised for a long time and may not work as indicated on the print form. Users
in need for more sophisticated printing options are welcome to contact me for an update of the
print functions. Till then I intend to leave things as they are...
4.1.14 e-mail current sequence
With this function you can send the currently displayed plain sequence by e-mail with an attached comment. In case you need to send the entire project the sequences must be saved in a multi-sequence file and e-mailed as an attachment using the standard e-mail Windows program.
4.1.15 close project and exit
Before SEQtools closes the user is adviced - twice - to save the project. Keep in mind
that SEQtools keeps all project data in RAM until the project is saved. Closing SEQtools without
saving the project will lead to irreversible loss of all data of the project.
Note that large batch blast search jobs - which may last several days - includes an option to
auto-save the project every time a specified number of searches has been performed. This reduces
the risk of data loss (in case of power failure for example) while the batch searching is
running. See section 4.4 of the manual for a more detailed description of this
option.
© 2002-2010S.W. Rasmussen (revised: )