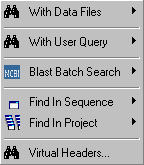
4.4 SEARCH MENU
- 4.4.1 about searching (general comments)
- 4.4.2 search with data files (construct plasmid)
- 4.4.2.1 compose restriction enzyme data file
- 4.4.2.2 restriction enzyme search, plasmid editor
- 4.4.2.3 restriction map
- 4.4.2.4 transfer selected region
- 4.4.3 search with user query
- 4.4.4 batch blast search
- 4.4.4.1 batch blast at genbank
- 4.4.4.2 local single database search
- 4.4.4.3 local multi-database search
- 4.4.5 find in sequence
- 4.4.6 find in project (patterns, identical, similar)
- 4.4.7 search virtual headers
 4.4.1 about searching
4.4.1 about searching
The search options in SEQtools are quite extensive. The brief descriptions below are primarily intended to give you an overview of the various options for searching sequences and their annotation. It is recommended that you look through the different sections below to learn which options are available. And then experiment to find the most convenient way to use the different functions.
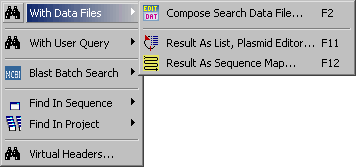
4.4.2 search with data files
This function enables you to sear a single sequence with a collection of restriction enzyme sequences.
Use Compose Search Data File to build the group of enzymes you want to include in the search data file. Note
that you can convert an entire project of primer sequences into a search datafile and use this datafile in exactly the
same way as proper restriction enzyme datafiles.
4.4.2.1 Compose restriction enzyme data file - This form contains various options for building a custom
designed restriction enzyme datafile. Use the setting User defined sequences if you wish to search with a
search datafile containing primer sequences (or other user designed search strings).

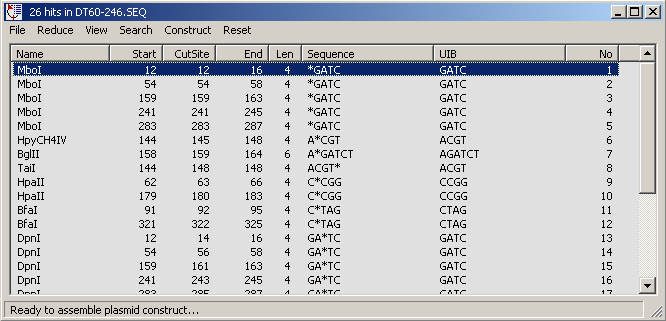
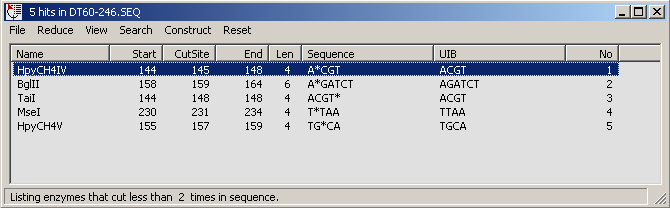
4.4.2.2 Restriction enzyme search, Plasmid editor - The results form shown below displays the result
of the datafile search. the form includes various options for filtering the list of matches (remove multi-cutting
enzymes for example). Clicking a match line highlights the match in the original sequence.
This form also contains a simple function for assembling simple plasmid constructs. This function is described in
the following screenshots.
Options under the Reduce menu.
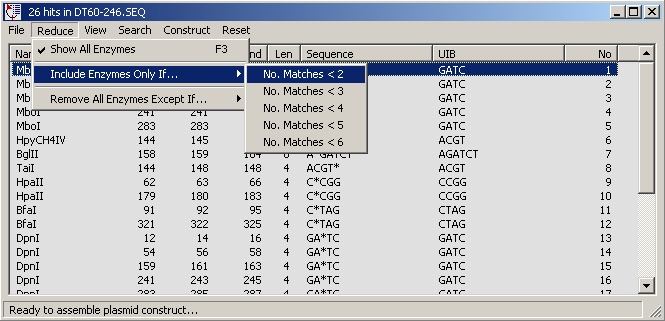
The match list after removal of restriction enzymes which cut more than once.
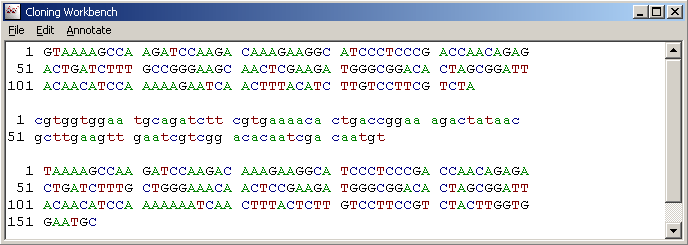
Proceeding through the steps of the Construct menu allows you to digest your sequence
with the specified restriction enzymes and to isolate the segment in a separate text form. This facility can
be used to build simple plasmid constructs: The operation comprises 3 steps:
(1) Navigate to the vector sequence in the project and isolate of 5' vector arm by a single cut.
(2) Navigate to the sequence to provide the insert and isolate the insert sequence by a double cut.
(3) Navigate back to the vector and and isolate the 3' vector arm by a single cut.
Navigating to a different sequence in the project automatically updates the list of matches for the selected
search data file. Before a sequence segment is inserted into the text form, the overhangs are checked for
compatibility and the result of the check displayed in an info message.
In the example shown below
the the 5' end of the construct is created by digesting the vector with enzyme HpyCH4IV. Clicking
Append 5' Vector Region To Construct transfers the isolated segment to a text editor.
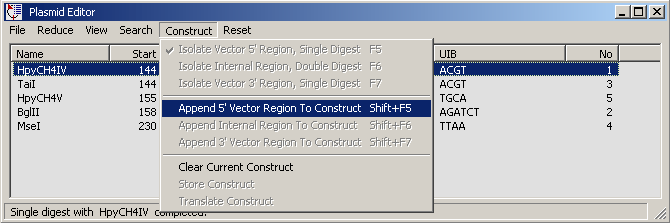
The 5' segment of the vector sequence after digesting with enzyme HpyCH4IV copied to the
text form.

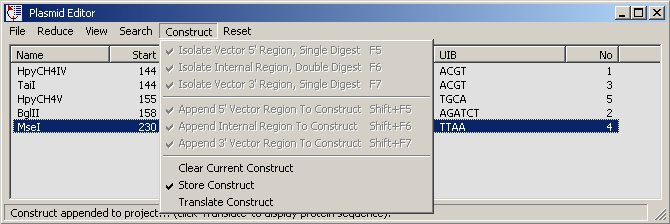
Isolating the insert by a double digest - hold down the <CTRL> key while clicking the second
enzyme in the match list. Note that this option is only available after the 5' segment has been
isolated.
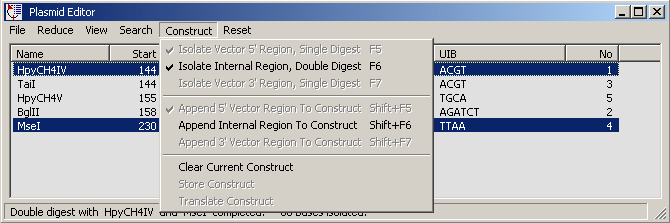
Click Append Internal Region To Construct to copy the isolated insert sequence to the text
form. In the screenshot below the construct has been completed by insertion of the 3' vector arm into the
text form.
The completed plasmid sequence can either be copy/pasted to a second nucleotide instance of SEQtools
or appended to the current project as a separate, new sequence file.
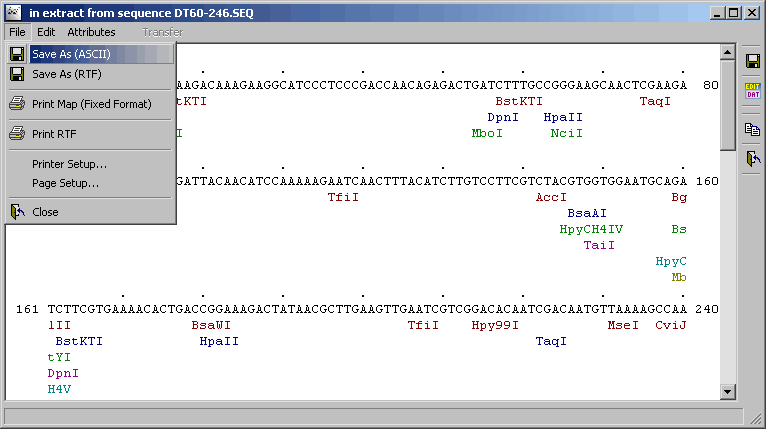
Restriction map - A restriction map is a second alternative for displaying the result of a
datafile search. The first character of the enzyme name marks the cut site.
4.4.2.4 Transfer selected region - Highlighting a region of the sequence in the restriction map and clicking Transfer closes the
restriction map form and transfers the highlighs to the sequence displayed in the normal SEQtools editor.
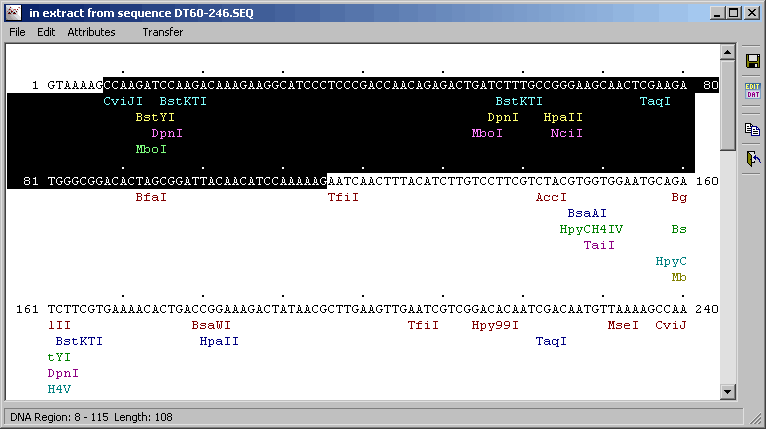
Highlighted sequence region transferred from the restriction map form.
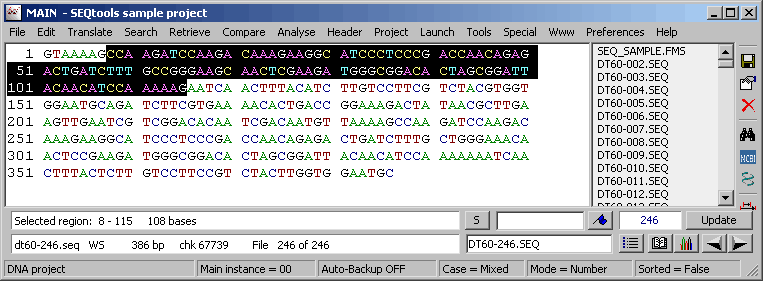
4.4.3 search with user query
Simple search with a sequence query (nucleotide or protein) can be performed either on the currently
displayed sequence or on all sequences contained in the project. In the latter case the Search Sequence
form automatically appears when a matching sequence is clicked in Project Search mode.
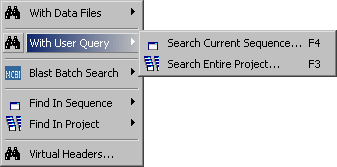
In addition to a plain query string more complex queries can be constructed
using the syntax below:
Syntax:
? Any character.
[ ] Any of the characters within the square brackets.
[! ] Any characters other than those within the square brackets.
5'/ABCn1-n2/ Between n1 and n2 characters from 5'-end or N-terminal other than A, B and C.
/ABCn1-n2/ 3' Between n1 and n2 characters from 3'-end or C-terminal other than A, B and C.
/ABCcn1-n2/ Between n1 and n2 characters other than A, B and C.
Examples:
Pattern: Finds: Does not find:
ASTS?V ASTSxV ASTSV
AST[GHWP]SV ASTGSV and ASTHSV ASTNSV and ASTRSV
AST[!GHWP]SV ASTKSV and ASTYSV ASTGSV and ASTHSV
/1-20/AST/4-8/SV 5' xxxASTxxxxSV 5' xxxASTxxxSV
AST/4-8/SV/2-20/ ASTxxxxSVxxx 3' ASTxxxxSVx 3'
/A1-20/AST/4-8/SV 5' xxxxASTxxxxxSV 5' xAxxASTxxxxxSV
AST/B4-8/SV/1-20/ ASTxxxxSVxxx 3' ASTxBxxSVxxx 3'
Where x is any character; 5' and 3' denote the 5'/N-terminal and 3'/C-terminal respectively.
Search current sequence - Result of sequence search. Each line include the start and end of the match
as well as the orientation (W=Watson; C=Crick) of the match.
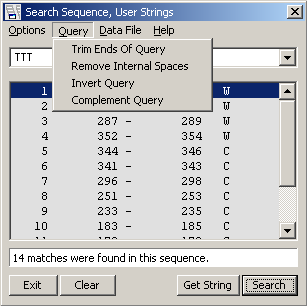
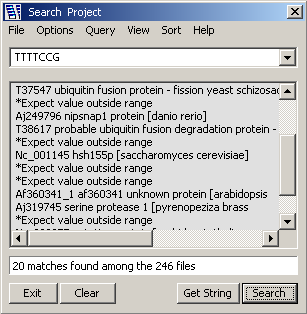
Search entire project - Result of a project search. The View option is set to Descriptions,
Virtual Blast Section listing the matching sequences by their best blast match in the blast search currently
selected in the Compose Header form as the Virtual Blast Search.
4.4.4 batch blast search
One the very strong features of SEQtools is the Batch Blast Functions allowing you to submit some or all sequences of a project to NCBI for homology searching og specified subsections of Genbank.
The Blast functions exist in two almost identical versions in SEQtools: One based on the QBlast scripts the other on the NCBI program blastcl3.exe. The first version is a web interface to the blast engine at NCBI while the other is a client/server type of arrangement.
In designing both functions a considerable efford has been spent on self-recovery of the functions in case of crashes to ensure that when a batch search job is launched it should run to completion without user intervention. This holds true in nearly all cases, even when the job lasts several days (TBlastX) or includes a large number of sequences (upto 30,000 has been searched successfully).
Results are nice provided you have somewhere to store them in a form that allows you to retrieve them again... The blast search functions of SEQtools are intimately integrated with the storage/retrieval system of search results. Read more about this under the Header menu.
Provided your pc is sufficiently powerful you can launch a batch blast job - and continue working (on a different project) in an different instance of SEQtools while the blast search runs in the background.
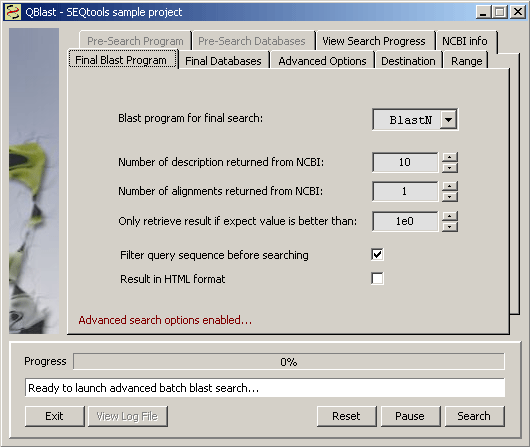
4.4.4.1 Batch blast at genbank - In most cases the different settings tabs are self-explanatory. Note, however, that you cannot import/parse blast results into sequence headers if you choose to get the results as html files. This has to do with the structure of the header/annotation. There is access to the Internet/Entrez at NCBI from within the sequence list displaying search results which to some extent compensates for this by providing an easy link to additional information.
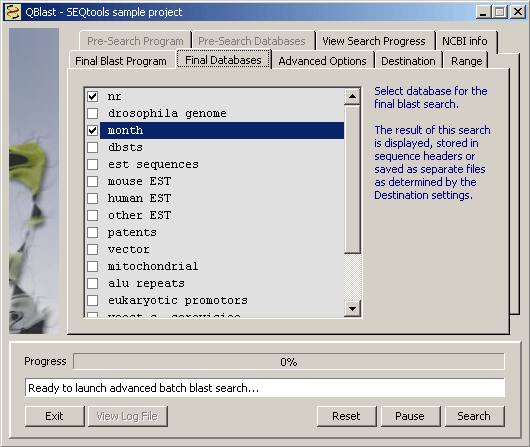
The list of available main sections of Genbank. The content of the database list (and the available blast programs in the dropdown list above as well) reflects the project type (nucleotide or protein).
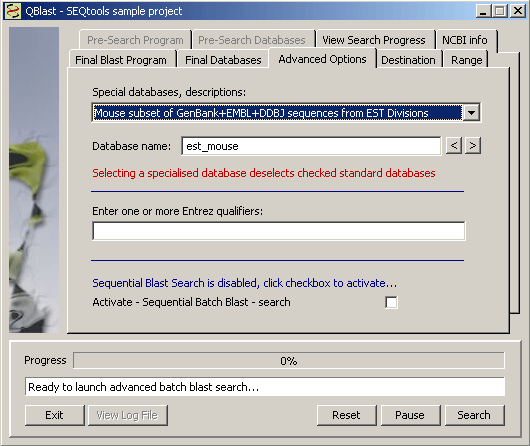
Under the Advanced Options tab is collected additional options for database selection. The list may not be entirely updated, but is the most recent the I could retrieve at NCBI.
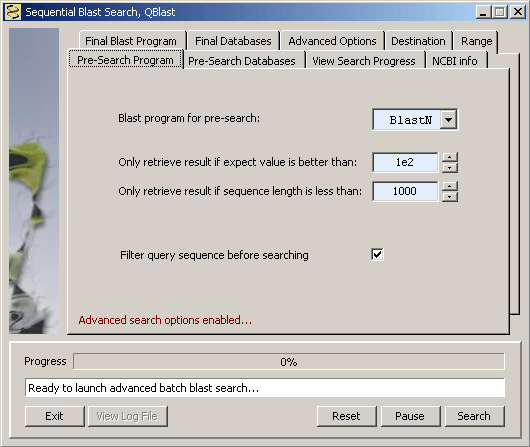
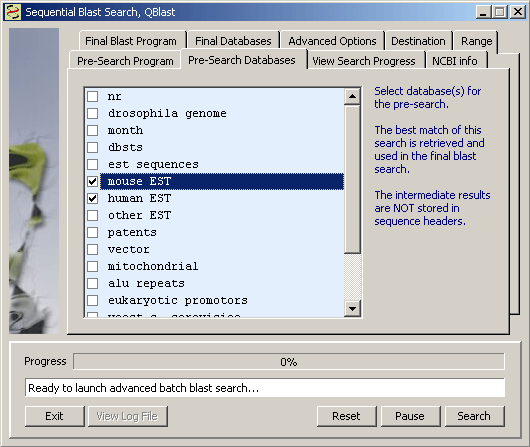
Among the advanced options is a checkbox for activating Sequential Search. This implies that the function performs two sequential blast searches: The first with the project sequences, the second with the best match of the first search. When this option is active two more tabs on the blast form becomes active to allow you to select program and database(s) for the first search.
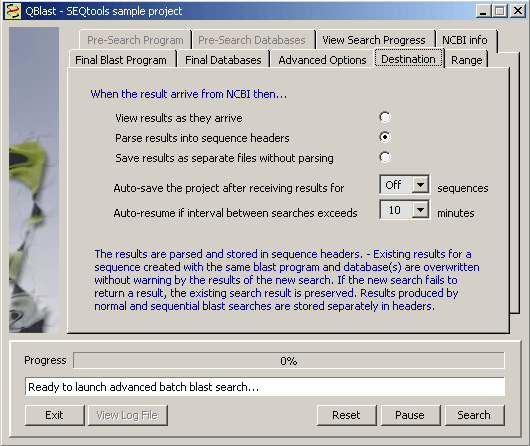
The Destination tab contains an option to save the project for the specified number of completed searches. As all data in SEQtools are stores in PAM until the project is saved this setting shouls be active and be set to for example save/100 searches. If the blast engine at NCBI is very buisy it may be an advantage to set the auto-resume value to 10 - 30 min to re-launch the sequence if more than the set amount of time has elapsed without a result has been received. Under normal circumstances it takes about 20-40 sec to search a 500 bp sequence with blastn.
You can choose to have the results displayed as they arrive in which case they are not stored in sequence headers. It is also possible to have the results saved as separate files (for example in html fornat). The default is Parse results into sequence headers.
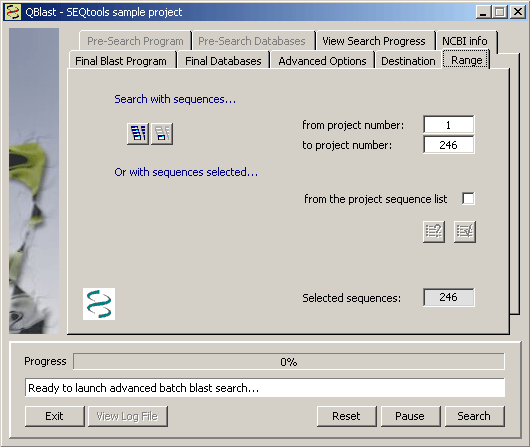
In this tab you can set the range of sequences you wish to search. This can be the entire project or the currently displayed sequence - or a discotinuous series of sequences selected from the project sequence list (described in detail on a separate page of the manual).
The tab for setting program options for the first search when Advanced options are enabled.
Available databases for the first search when Advanced options are enabled.
4.4.4.2 Local single database search - This function is for searching the project sequences with a local database, created by the user. Locak databases can be created in different ways as described under the Tools menu. When you launch the local database search form, ao message box (see below) informs you about available databases and displays a link to the function used to create local databases. The results are stored in sequence headers in exactly the same way as results from searches at NCBI.
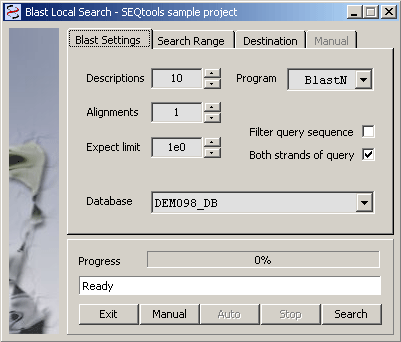
The message box informing you about available local databases.
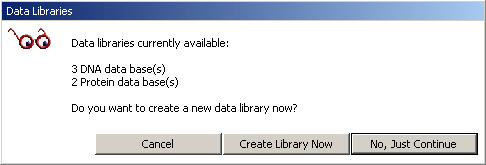
To perform a Manual search, just highlight a region in the displayed sequence and click Get Seq to import the query into the local blast search form. Click Search to run the search.
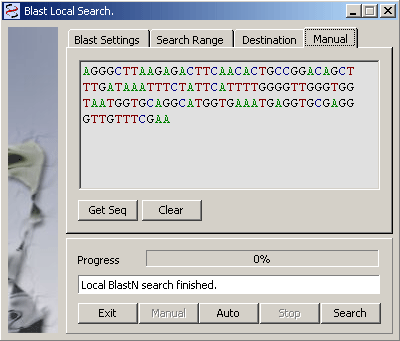
4.4.4.3 Local multi-database search - Occasionally you may want to perform a local search on more than one local database. This can be achomplished with the Search Multi-Database function. Settings are the same as for other blast functions except it is possible to select more than one local databases.
Note that it is not possible to store more than one multi-database blast search in the sequence header. Running a second search overwrites the first one without warning. You shoutd consider this function primarily as a help assisting you in getting an overview of the project rather than a proper analysis of individual sequences.
Clicking a line retrieves the selected sequence/sequence header (if displayed). You can the use the facilities (Compose New Project) in the sequence Header form to isolate interestering sequences into a separate project - simply by selecting and clicking.
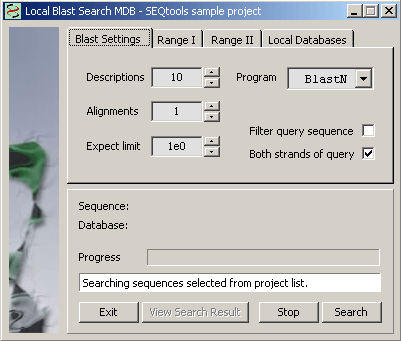
Selection of local databases for a local multi-database blast search.

Setting the search range. The range can either be the displayed sequence or all project sequences (Range I) or a discountinuous series of sequences selected from the project sequence list (Range II).
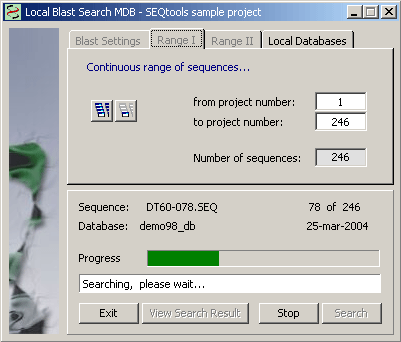
The results of a multi-database blast search is arranged somewhat differently in the sequence headers. All search results for a given sequence with the selected databases are contained in a single section of the header.
To create an overview of the multi-database search results the form shown below retrieves and displays the best multi-database search results for all sequences of the project.
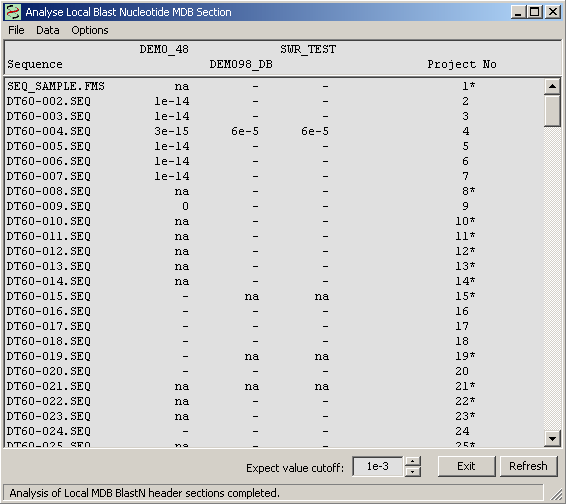
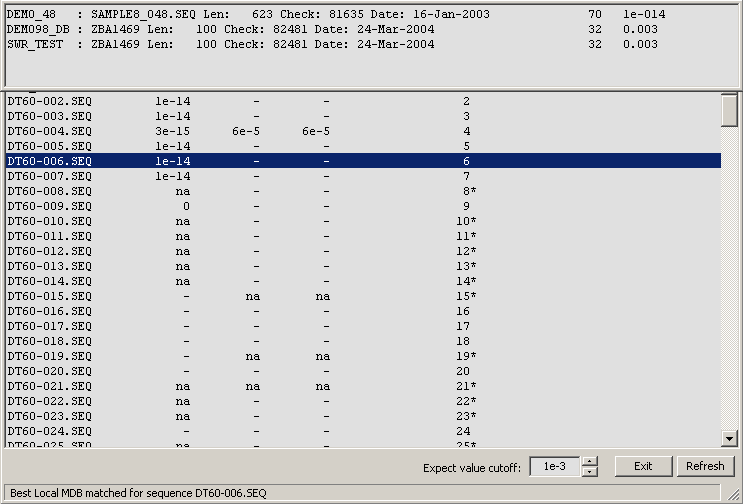
Highlighting a line in the form and then holding down the right mouse button retrieves the best match for all databases for the selected sequence.
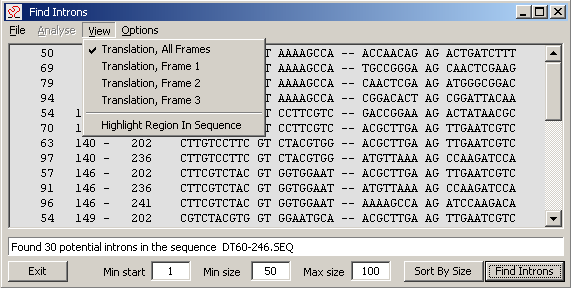
4.4.5 find in sequence
Two are included for revealing the existance of repeats in the sequence and for detecting/indicating the presence of introns (primarily in yeast). Neither function should be considered as perfect. Much more sophisticated functions are required to identify introns in mammalian genes and the user is strongly adviced to visit websites specifically directed towards this analysis.
![]()
4.4.5.1 Repeats - Identifies direct and inverted repeated regions in sequences.
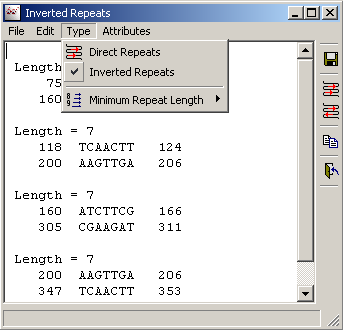
4.4.6.2 Introns - Primitive function for identification of introns in yeast.
4.4.6 find in project
The four functions described below are all designed to perform analyses on the entire project. This includes finding duplicate sequences, performing a quick project blast search to reveal internal similarity among project sequences, a emboss based pattern search and finally a function to identify project sequences with antisense blast matches to sequences in sequence header.
4.4.6.1 Duplicates - Scans project and lists duplicate sequences. Duplicate sequences can then be selected and removed from the project.

4.4.6.2 Project blast - Project blast search builds a local database (if not already present) and performs a blast search of the currently displayed sequence against the project database.
The result is either displayed in a simple text form (below) or in the sequence list (with light blue background) of the main sequence editor of SEQtools. The latter display can be achieved by clicking <F5>.
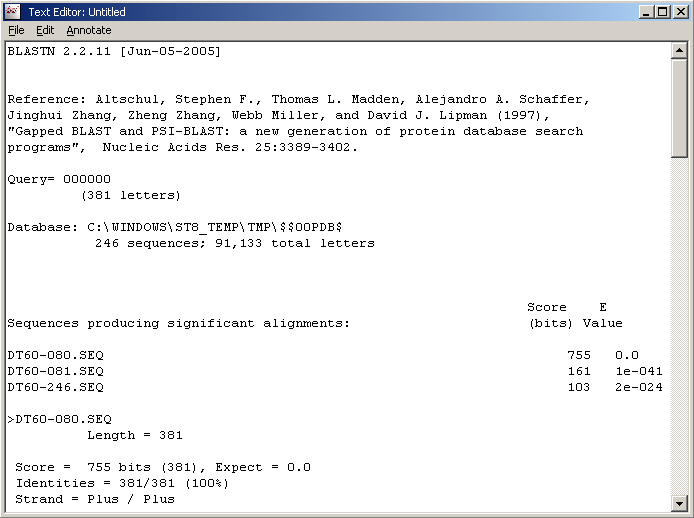
Project blast results for displayed sequence. Right-click the sequnece list to return to the normal list (grey=load order or yellow=sorted).
The list of project blast matches is linked to an alignment function (ClustalW): Select some or all matches (click while holding down <CTRL>) and Shift-Right click to open the popup menu offering to access to project blast preferences, Close Selected Sequences or Align Selected Sequences as illustrated by the screenshot below.

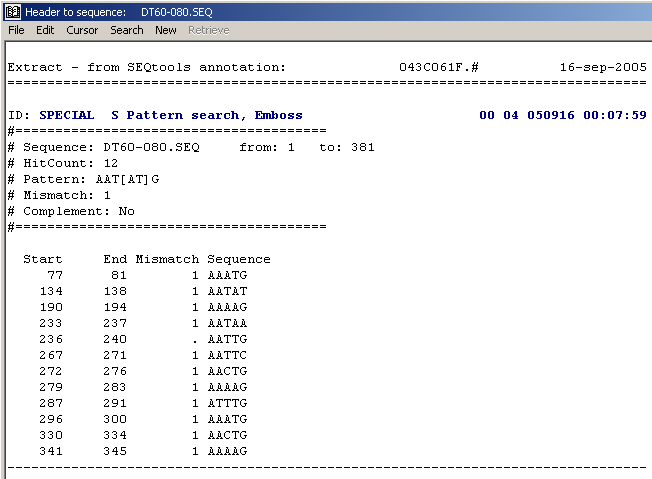
4.4.6.3 Patterns - The pattern search function utilises emboss functions to find patterns in project sequences. The Range settings are the same as for other search programs in SEQtools.
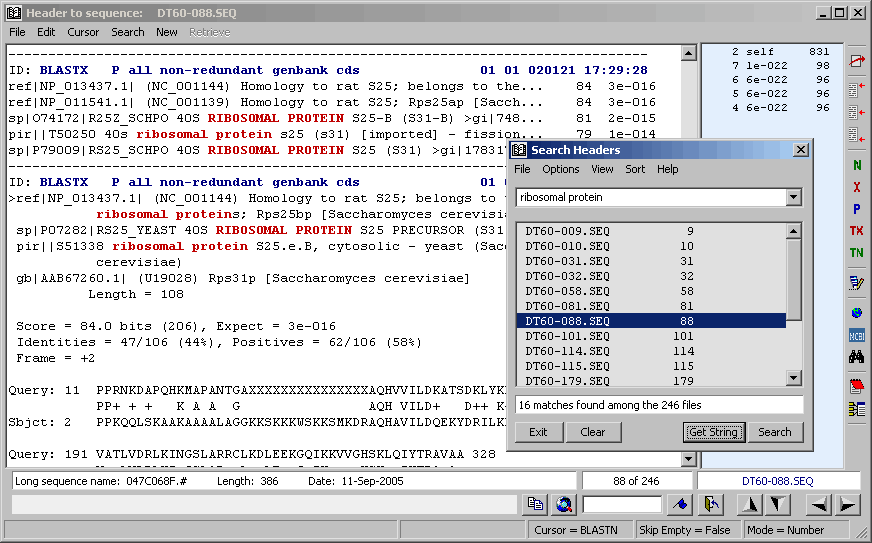
The syntax is briefly described in the form below (consult the emboss homepage for additional details). The result of the pattern search is stored in the sequence headers and are displayed by clicking the View Header command button.

The sequence header displaying the results of a pattern search. As for local multi-database blast search it is only possible to store the results of a single pattern sear. The next search will overwrite the existing results without warning.
Setting the Range parameter for a pattern search.
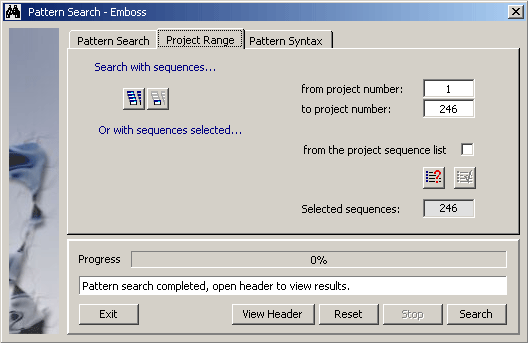
The brief description of the pattern syntax. Consult the emboss homepage for details and additional examples.

4.4.6.4 Antisense - This function searches all headers of the project and examines the alignment sections of blast results (if present) for the selected Virtual Blast Search and lists the strand orientation for the best match of each sequence in the project. It is possible to select project sequences to be complemented to make the orientation of the sequence and its database match the same.

4.4.7 search virtual headers
The last search function enables you to search sequence headers with plain text queries. The menu options include case sensitive/insensitive, whole word only, match listing as sequence data or descriptions.
Search headers - match listing by sequence data.
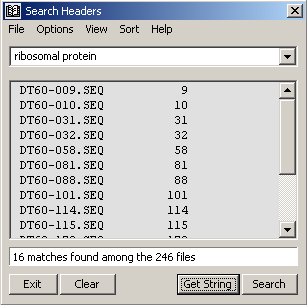
View setting set to Descriptions, Virtual Blast Sections causes match listing by description lines.

Clicking a line in the header results form retrieves the relevant sequence header and paints matches to the query string red in the header text. Note that the header search is limited to the currently selected items (checkmarked in the Compose Header form).
© 2002-2010S.W. Rasmussen (revised: )