4.5 RETRIEVE MENU
- 4.5.1 about retrieve functions (general comments)
- 4.5.2 auto-annotate project
- 4.5.3 new project from genbank records
- 4.5.4 convert genbank ac to gi
- 4.5.5 entrez
- 4.5.6 locus link
- 4.5.7 import annotation from ms-file
 4.5.1 about retrieve functions
4.5.1 about retrieve functions
In addition to annotation by blast searching SEQtools contains several functions designed to automatically retrieve sequence annotation from various sources.
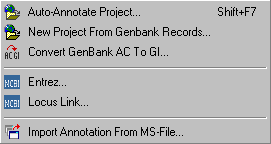
4.5.2 auto-annotate project
With this function you can automatically retreive sequence annotation (Genbank records and/or Medline) for the blast search currently selected as the Virtual Blast Search. As in batch blast functions you can set the sequence Range to current, all or selected group.
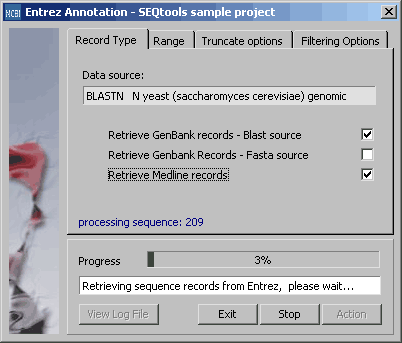
Tab for setting the range parameter for retrieval.
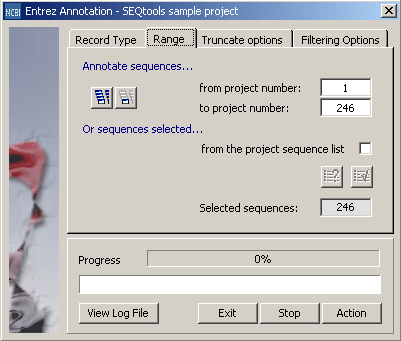
Tab for setting lower limit (maximum expect value for retrieval of annotation) for retrieval. The second parameter tells the retrieval function to preferentially retrieve Medline information from the specified Genbank subsection (ALL, REF, DBJ, EMB, GB or GI) - but only if the expect value of the match is within the specified cutoff limit (in percent of retrieval expect value cutoff).
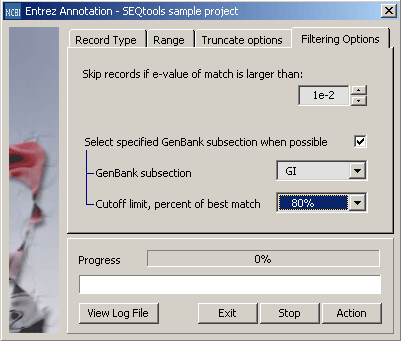
4.5.3 new project from genbank records
A rather complicated function which allows you to create an entirely new SEQtools project composed of sequences retrieved (best matches of one or more specified set of blast results) from Genbank.
The firet step is to select the blast searches to include in the retrieval process. The list of available blast searches is prioritised and can be modified by the Up, Down and Remove buttons. Once the list is finished, click the Build New Gene List tab.
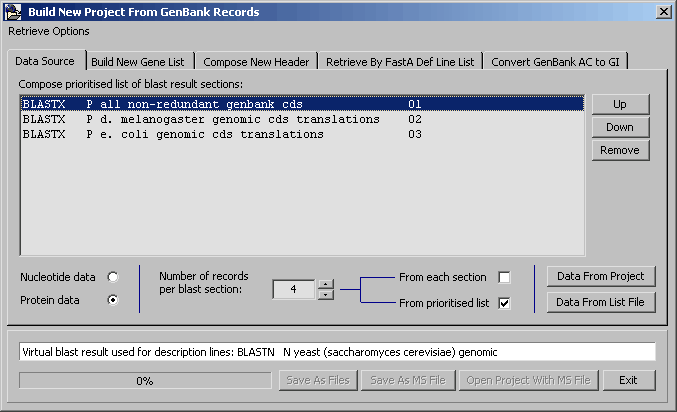
Clicking the Build List causes SEQtools to scan all header of the project for the information required to retrieve sequences from Genbank. The retrieved information is displayed in the sequence list. Use Remove Selected and Remove Duplicates to clean the list of sequences to be retrieved from Genbank.
Clicking Retrieve Records launches the retrieval process.
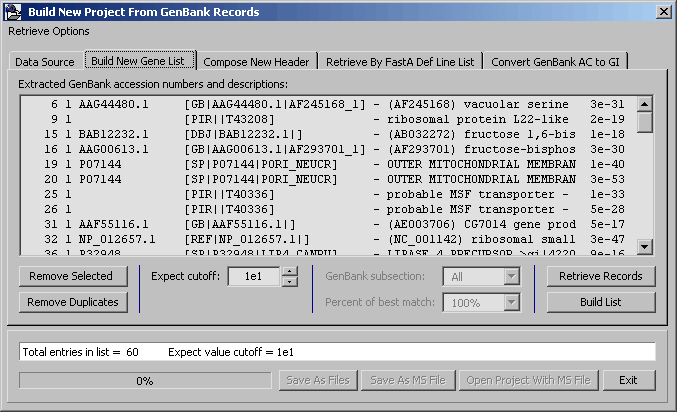
SEQtoold then downloads the selected sequences from Genbank. The list below displays the progress (and rejected sequences). The list also includes a short extract of the retrieved sequence.

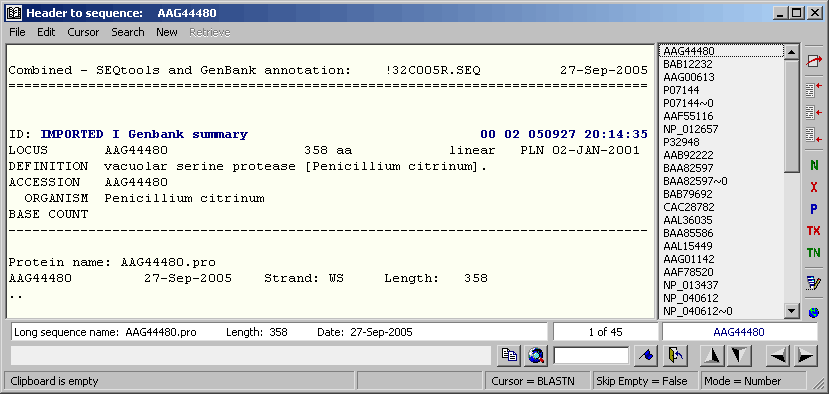
When downloading is completed the Compose New Header tab is displayed. In this tab you can select information to be transferred from the current to the new SEQtools project.
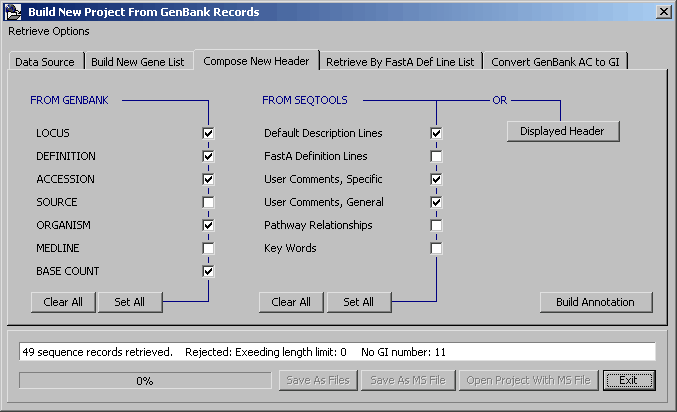
Finally click Build Annotation and save the new project as a SEQtools formatted multi-sequence file. Clicking Open Project With MS-File launches a new instance of SEQtools containing the Genbank sequences derived from the original project.
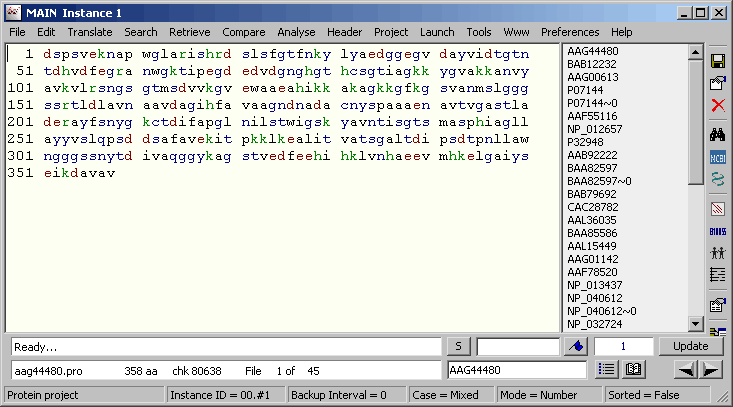
The sequence annotation transferred from the original project to the derived Genbank project.
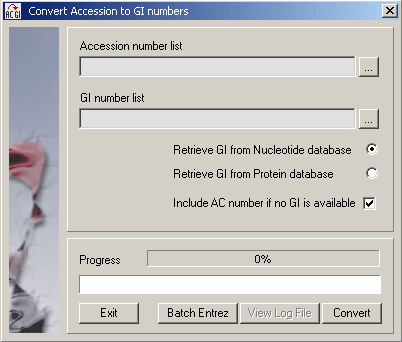
4.5.4 convert genbank ac to gi
A simple translator converting a list of accession (AC) numbers to a list of GI numbers.
4.5.5 entrez
"The life science search engine" - the key source of biological information at NCBI.
No description is necessary, visit the site and see for yourself...

4.5.6 entrez gene (locus link
)
The old LocusLink site has been is superceded by Entrex Gene, a searchable databa of genes,
fromRefSeqgenomes, and defined by sequence and/or located in the
NCBI Map Viewer.
4.5.7 import annotation from ms-file
If information relating to the same set of sequences but present in two different SEQtools projects (i.e., with exactly the same sequences) it is possible with this function to import annotation from a second project containing the same sequences (but with different information stored in sequence headers). Simply choose the SEQtools formatted multi-sequence file and select the information you wish to import.
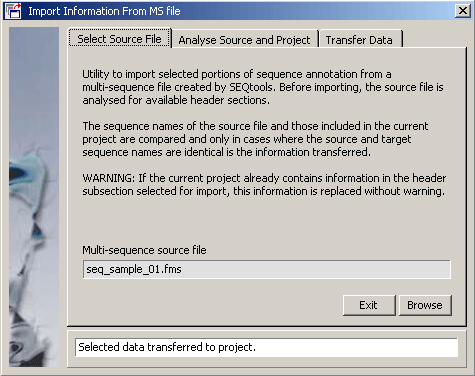
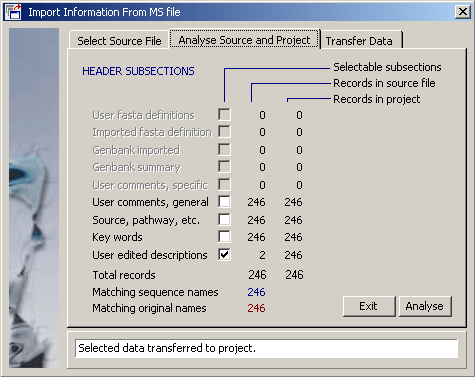
In this example various user created information is available in the multi-sequence file.
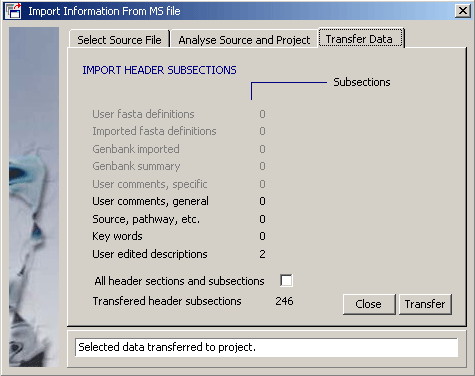
Clicking Transfer starts the import process, listing the number of imported header sections. Remember that nothing is permanent in SEQtools until the project is saved. There is no Redo option here so simply close the project without saving it to cancel the operation.
© 2002-2010S.W. Rasmussen (revised: )