4.11 TOOLS MENU
- 4.11.1 about tools
- 4.11.2 translators
- 4.11.3 conversion functions
- 4.11.3.1 convert project to data file
- 4.11.3.2 accession number to GI converter
- 4.11.3.3 restriction enzyme data file converter
- 4.11.4 multi-sequence functions
- 4.11.4.1 create local databases
- 4.11.4.2 multi-sequence tools
- 4.11.4.3 build project from genbank records
- 4.11.4.4 import annotation
- 4.11.5 file tools
- 4.11.5.1 search and view text files
- 4.11.5.2 lifetrace data file viewer
- 4.11.5.3 primer order file viewer
- 4.11.5.4 file and folder tools
- 4.11.6 editors
- 4.11.6.1 sequence merge editor
- 4.11.6.2 text editor
- 4.11.6.3 search data file editor
- 4.11.6.4 fasta definition line editor
- 4.11.7 calculators
- 4.11.8 create files
 4.11.1 about tools
4.11.1 about tools
The Tools menu groups a series of functions for special purposes. Some of the tools functions
are also included under other SEQtools menus but are repeated here to facilitate accessing/locating them functions.
Look through the list below to get acquainted with the options available by these tools.
4.11.2 translators
Simple translators included to the benefit of users of SEQtools (including myself) who still have to
look in a test book to find the amino acid corresponding to a particular codon and who have problems remembering
the IUB symbols (published by Commission
on Biochemical Nomenclature) for degenerate positions in sequences.
![]()
4.11.2.1 IUB symbol converter - Symbol Translator for conversion of degenerate bases of a
codon to and from the standard IUB symbols (Nomenclature Committee, 1985, Eur. J. Biochem. 150, 1-5). These symbols
are used by the EMBL, Genbank and PIR data libraries and are recognized by the programs of the GCG
package.
Translate from IUB symbol to Normal syntax.
![]()
Translate from Normal syntax to IUB symbol.
![]()
4.11.2.2 Codon calculator - The Codon Calculator is used to translate codons into amino acids or
amino acids into their respective codons. The list of valid codons for a particular amino acid also includes the
corresponding degenerate codon using the IUB symbol table.
The tab key or a click toggles between codon
and amino acid input. N's are allowed when entering codons. The function of the Codon Calculator is
illustrated in the screenshots below.
![]()
![]()
![]()
4.11.3 conversion functions
Three functions for conversion purposes. The first converts an entire project into a search
data file which can be used in the same way as restriction enzyme data flies. In most cases this would be primer
sequences for PCR or sequencing purposes. The remaining two converters reformats a restriction enzyme data file
downloaded from ReBase in GCG format and
converts gene accession numbers (AC) to GI numbers.

4.11.3.1 Convert project to data file - Using SEQtools as a primer "database" including the
primer sequences in a normal nucleotide project offers several advantages: The primer sequences can be
annotated/verified by Genbank searching relevant databases. This annotation is stored in the sequence header
of the primer sequence. You can then use sequence listing and searching facilities to identify for example
primers for a particular organism, gene etc.
With this function all sequences of the current project can be converted into a SEQtools search data file.
This is especially useful to verify the sequence and location of primer sequences (sequencing and PRC primers)
derived from a known sequence.
The *.sdf file can be used to search project sequence in exactly the same way as normal restriction enzyme
search data files. Displaying the primers in a map or as a list, just as described for restriction sites
greatly facilitates the verification of the sequence and location of primers.
Note that the Convert Project function uses the exact same sequence as included in the project.
This implies that restriction sites or linkers which may have been added to the primer sequence are included
in the search data file.
4.11.3.2 Accession to GI number converter - Submits a list of genbank accession numbers (AC) to entrez
to get the corresponding GI numbers.

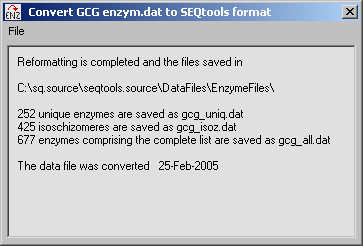
4.11.3.3 Restriction enzyme data file converter - A small program used to convert the restriction enzyme
data files in GCG format to SEQtools format. Further editing of the converted data file can be carried out with the
data file editor (see below).
To convert the data file simply download the restriction enzyme data file in GCG format from
Rebase and follow the instructions under
the File menu of the Converter form. Note that in addition to enzyme data files, ReBase contains
very useful search function which allows you to search their data base, either with the name of an enzyme or
with a recognition pattern.
4.11.4 multi-sequence functions
The first item on the, Create Local Databases menu is described is some detail. It is my experience
from many user comments that this essential feature of SEQtools is sometimes difficult to get to work. The next menu
item, on the other hand, contains a collection of old routines whish are seldom used. The current descriptions are copied
from the previous manual and not check very carefully here. New
Project From Genbank Records and Import Annotation From
MS-File are also included under Retrieve menu and are only described very briefly here.

4.11.4.1 Create local databases - This form contains several functions and utilities for creating local
database files. Building a local database involves two steps: (1) Create a FastA source file for formatdb
(routine provided by NCBI to build blast searchable databases). (2) Run formatdb with the source file
just created.
This collection of programs allows you to (1) combine all sequences located in a particular directory, (2) all
sequences included in a SEQtools project or (3) sequences you type yourself into a source file for formatdb.
The form also includes a simple tool for managing existing SEQtools databases which is handy if you want to
remove a databases from the list.
(1) Create source files from files in a directory - to create a FastA source file it is necessary to collect
all the sequences you wish to include in the source file in the same directory. Remember ALL files in the
specified directory are included in the database - also text and program files not containing sequences -
so please check the content of the directory carefully before you start building the database.
The File and Folder Tools utility described below is useful in getting a list of the files to be included
in the database. If the sequences are annotated by SEQtools it is also necessary to indicate which header section
you want to use for annotating the source file and thus also the database.
(2) Build source file from sequences in a SEQtools project - This function works in the same way as described
above, except that the FastA source file is built from the sequences and their headers contained in the current
SEQtools project.
To create a local database simply select the source file you wish to convert, choose a name for the database and
select whether the source file contains nucleotide or protein sequences. Check the -o option enabling later retrieval
of sequences from the database. Then click the Action command button.
The files comprising the local database are created in the DT8_TEMP/DB/ folder in the Windows/Winnt folder - and must
remain there if you wish to search it. A copy of the database is stored in ...\SEQtools\_databases.
In case you want to create a protein data base from a project containing DNA sequences use the Create Protein Files
under the Translate menu to translate the nucleotide sequences into protein before building the source file and
the final data base.
(3)User strings databases - In case you wish to create a database from a smaller number of sequence motifs,
you can use this function to enter nucleotide or protein strings, give them a name each and finally convert the
strings into a searchable database.
The sequences included in the
database will contain only a single annotation line. If the input sequences are in Genbank, FastA or GCG
format, the annotation is automatically retrieved from the original files. For Genbank files the DESCRIPTION line
is used, for FastA, the standard, single line annotation is included and for GCG, the line immediately before
the .. divider.
For SEQtools files, the header sections listed on the form select which header section is used for annotation. The
default is sequence name, length, check sum and file date. Obviously only header
sections actually containing information can be used to annotate the database.
After a while, the number of local databases may become too large making it
difficult to find the one you wish to use. With this facility you can remove databases from the list. There is
no undo option so the operation is final. It is, however, possible to rescue a deleted database by
locating the database files generated by formatdb in the ...\SEQtools\_databases folder and moving them to the
general DOS folder, specified above.
The most important tabs of the Local Database Tools form are
shown below:
The Build FastaA Source File tab.
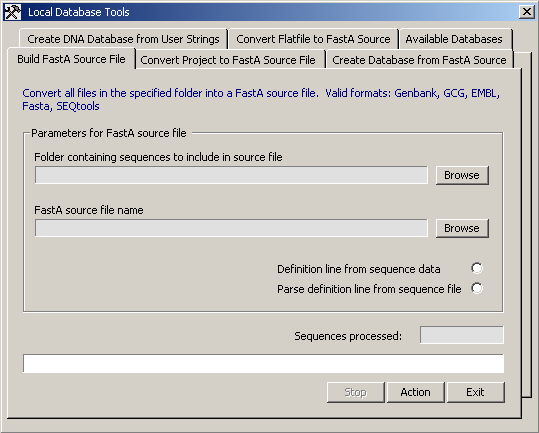
The Convert Project to FastA Source File tab.
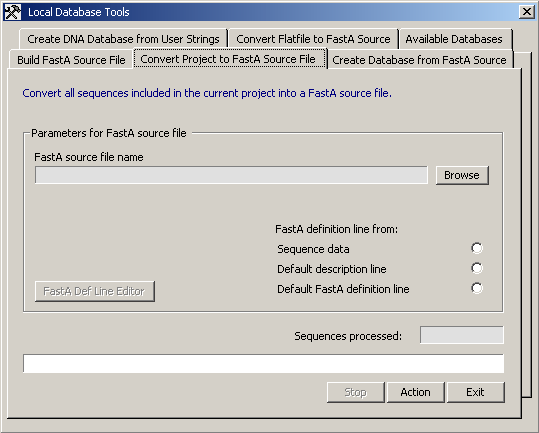
The Create Database from FastA Source tab.

4.11.4.2 Multi-sequence tools - This function allows you to modify or create multi-sequence files. Database
files in FlatFile or FastA format consist of text documents containing multiple database records in plain ASCII
text format and can be downloaded from several web sites. The description below is the unmodified from the previous
SEQtools web manual and may not be correct in all aspects.
Format multi-sequence files - This function checks the EOL (end of line) codes of multi-sequence files to
see if LF is used alone. If this is the case, all LF codes are replaced by CRLF codes and the formatted
file is saved with the same name but with the extension *.fgf (Formatted Genbank File). In cases where the correct
CR code is used, a copy of the file is saved with the *.fgf extension.
The Process Annotation functions reduce the annotation of each of the sequences in a Genbank flatfile
or a collection of Genbank files located in the specified folder leaving only the DESCRIPTION line and the ACCESSION
line intact. The ORIGIN divider and the // record separator are also maintained in the trimmed file.
Sequences contained in a multi-sequence file - If a value is entered in the reject field, sequences longer
than the specified length are excluded from the trimmed file. In most cases such long sequences comprise
multi-gene sequences which may be undesirable.
Separate sequence files in a folder - The input file must be a file with the *.fgf or *.ngf extension, i.e.
a user generated file or a Genbank file where the EOL codes have been checked and replaced if incorrect.
The trimmed file is saved with the same name as the input file but with the extension *.tgf
(Trimmed Genbank File).
Derived genbank - This function allows you to extract a subgroup of sequences from a formatted Genbank file,
e.g. EST sequences derived from a specific organism from the complete EST data base flat file. The Genbank file must
be in standard format where the sequence portion of each record is delimited by ORIGIN and //. The annotation part
must include DEFINITION , ACCESSION, ORGANISM and REFERENCE sections. This function does not work with multi-sequence
files in FastA format or with trimmed Genbank files.
Criteria - The criteria for selecting files for the derived Genbank file are entered using the logical operators
AND and NOT as exemplified below:
- AND Homo (retains all files containing the word Homo/HOMO/hoMo in the annotation
- NOT Yeast NOT Saccharomy NOT cerevisiae (selects all sequences except yeast sequences)
- AND Plant (retains all plant sequences)
- AND Fungi (retains fungal sequences)
Note that a maximum of five AND and five NOT words are allowed in the criteria string. If more than five
words in each category are included, the excess words are disregarded in the selection. The logical operators and the
keywords must be separated by spaces. The search is case-insensitive.
The input file must be a file with the *.fgf or *.ngf extension, i.e. a user generated Genbank file or a Genbank file
where the EOL codes have been checked and replaced if incorrect. The derived file is saved with the same name as the
input file but with the extension *.dgf (Derived Genbank File).
Obviously, this function is only relevant for Genbank files where the annotation has not been trimmed with the Remove
annotation function.
Create FastA flatfile - With this function it is possible to build a new flatfile formatted Genbank file from
your own sequences. Before you attempt to build the file, make sure that all sequences to be included in the file are
located in the same directory. The record format of the new Genbank file is almost the same as that of trimmed Genbank
files and the extension of the new file is *.ngf (New Genbank File).
The only sequence identifying information preserved is the file name which is included after DESCRIPTION, all other
information contained in the header is lost.
SEQtools accepts files both in Genbank, SEQtools and FastA format. In some cases, where entire data base files
are present in the file header of files to be included into a Genbank flatfile, more than one header/sequence
divider may appear in the file. This will result in incorrect separation of header/annotation and sequence.
The solution to this problem is to load the files into SEQtools and save them again: SEQtools checks each file header
for illegal dividers in the header before the file is saved. If multiple dividers are encountered in the header,
they are converted to characters not recognized as dividers.
This function can be used to create a multi-sequence file suited for sage tag extraction. In other words, you can
avoid loading large numbers of files into a project for sage tag extraction if, instead, you first combine them
into a multi-sequence file and then use the sage extraction function for Genbank files to generate the sage
tag file.
Break-up multi-sequence files - This function breaks-up a multi-sequence file in Genbank or FastA format and
saves each sub-file in the same directory. The file names of the sub-sequences is composed of the accession number
of the sub-sequence with the extension *.gbk for Genbank files and *.fsa for FastA files.
4.11.4.3 Build project from Genbank sequences - This rather complex function for creating a new SEQtools
project from downloaded Genbank records is explained in detail under
the Retrieve menu and will not be described here
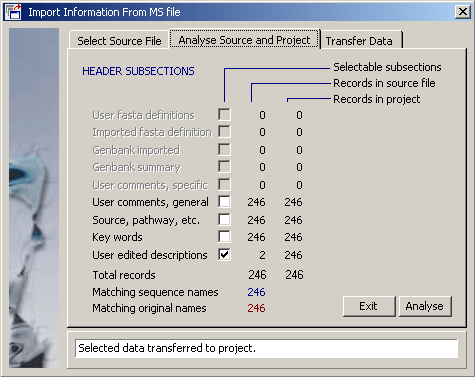
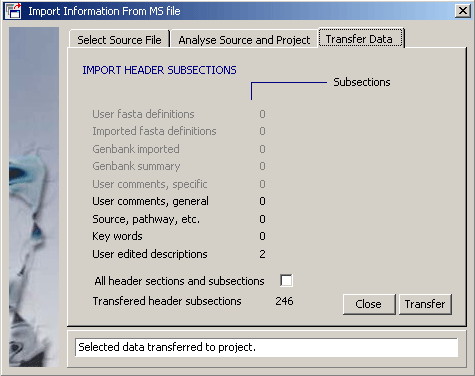
4.11.4.4 Import annotation - The function for importing user created sequence annotation from a multi-sequence
SEQtools file into the current project sequences is described under
the Retrieve menu and will not be explained here.
4.11.5 file tools
A group of functions for performing various tasks on plain text files and
SEQtools generated data files.
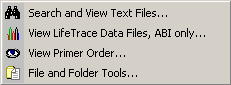
4.11.5.1 Search and view text files - Utility for viewing and searching all text files located in the
specified directory without having to open the files manually. After selecting the directory and a search string, all
files are opened - one by one - and the content automatically displayed.
The search string can include three logical operators: AND, NOT, OR. Note that these operators
will not work as parts of a search text string. The logical operators must be included by spaces to be
recognized.
Example: Protein AND Gene NOT Ribosome OR Chloroplast
If a search string is not entered, all selected files will be displayed and included in the match-group. In the
View menu you can select which files to display during the search: None, Matches, No matches
or All.
When the search is completed, double clicking a file name in the Match or the No-Match panel will
retrieve and display the content of the file. The displayed file can be printed from the Printer Options
menu, or by pressing <CRLF+P>.
The original files are not modified by this function.
4.11.5.2 Lifetrace data file viewer - The basecaller LifeTrace is an advanced external basecaller
for processing chromatograms generated by automated sequencers. The LifeTrace program and its
setup is
described in detail under the File menu. When debugging
this program and its basecalling it
is sometimes convenient to be able to view the data files (qscores, q and
gap_qscores) in a slightly
more organised way.


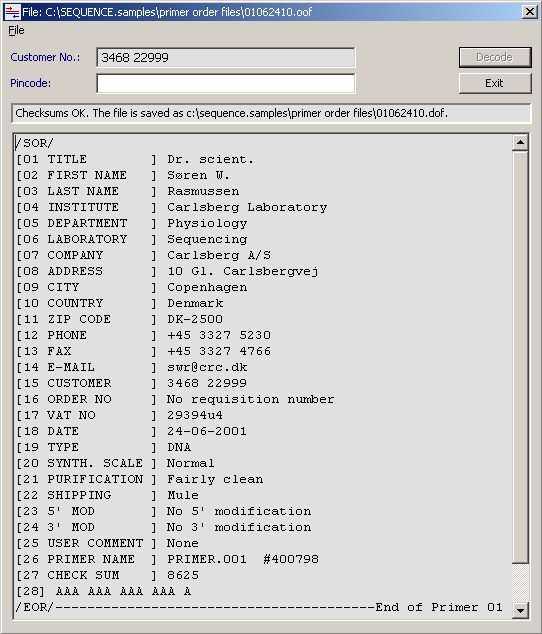
4.11.5.3 Primer order file viewer - If you are using SEQtools to handle your primer ordering, this
viewer help you inspect primer order files. If you are using the encryption facility, the pincode used when
saving the order file must be entered when viewing the file later. Read more about SEQtools and primers for
sequencing and PCR on the separate page for Primer
Design.
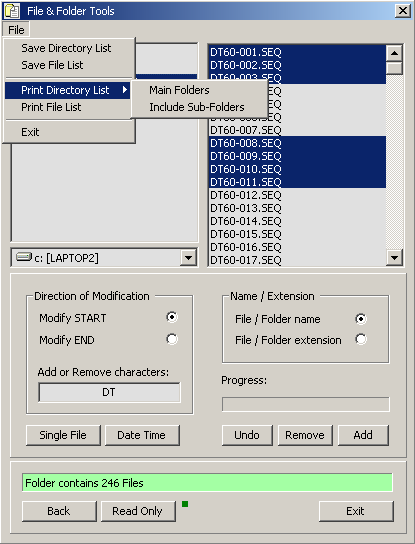
4.11.5.4 File and Folder tools - This small program enables you to carry out a number of operations
on file and folder names. You can edit the file titles and extensions, change file dates, print and save file
and folder lists etc.
The program is very useful if - for example - you want to print out an index of the content of a CD or change
all file dates to the current date.
4.11.6 editors
This menu contain four editors for various purposes. The sequence merge editor is described under
the Compare menu and will not be described further
here. The Text Editor is an simple editor similar to Notepad. The data file editor is a utility for creating and
editing different types of search data files. The FastA definition line editor is a more complex editor to enable
you to customise definition lines for saving project files in FastA format.
4.11.6.1 Sequence merge editor - This function is designed to assist you in building and editing merges
of overlapping nucleotide sequences. Although this merge function appears rather primitive, extended use of the
function for finishing small sequencing projects has demonstrated that it works quite effectively in creating an
error-free consensus sequence from the sub sequences.

4.11.6.2 Text editor - A simple text editor for writing and saving plain text files. In addition to
plain text, the text editor loads and saves files in rich text format.
The text editor provides a number of properties you can use to apply formatting to any portion of text. To change
the formatting of text, it must first be selected. Only selected text can be assigned character and paragraph
formatting. Using these properties, you can make text bold or italic, change the color, and create superscripts
and subscripts.
4.11.6.3 Data file editor - The data file editor is used to make corrections and additions to existing
search data files *.sdf or *.dat . In addition, the you can create new data files containing various collections
of search strings such as selected groups of restriction enzyme recognition sites, DNA or protein motifs, primers,
PCR primers etc.
By default, the currently active data file is loaded into the editor when it is opened. To choose another data file
for editing - or to create a new one use the File menu. Depending on the selection, the form changes slightly
to accept DNA or protein search strings. Before a new entry can be appended to the data file file, the string is
tested to see if...
- a search string name has been entered - and that the name is not already already in the data file.
- a search string has been entered in the text field - and that the string is not already in the data file.
- cutting sites have been correctly entered in the Watson and Crick strands (DNA sequences only).
To add a new item to the data file type the name and the search string in the respective
fields and click Append.
Cleavage sites - A star * is used to denote the position of a cleavage site in the Watson strand and
a <space> in the Crick strand. of DNA sequences.
Valid characters - The following characters A , C, G, T, N, * and <space> are allowed for restriction
enzyme sequences. Deleting single characters can only be bone by the backspace key from the current position until
the character is removed. All other editing keys are deactivated.
For protein and DNA motifs other than restriction enzyme sequences, all keys are active and there are no l
imitations - or verification - of the search string. See the detailed description of complex search patterns
under the Search menu.
Crick strand - The sequence of the crick strand is generated automatically and cannot be edited.
Maximum length - The maximum length of a search string is 100 characters. If cut sites are omitted, the
string is classified as a user defined string.
Validation - Validating the entry evokes the validation
function without appending the search string.
Sort Data File - This option retrieves the complete data file, sorts it alphabetically and saves the
sorted file. Optimizing the data file in this way improves the performance of the search facility of SEQtools.
A copy of the original file ($ replacing the last character of the file name) is saved before the
sorted file.
Convert To Complement - If this checkbox is checked, the currently displayed sequence are converted to its
complementary strand (only for restriction enzyme sequences).

The Edit menu of the data file editor.

4.11.6.4 FastA definition line editor - Utility for composing definition lines for sequences to be saved
in FastA format. To use this function, it is necessary that all files to be saved in FastA format are loaded into a
SEQtools project. As the function utilises the header information of the sequences, it is also necessary that the
sequences are properly annotated, i.e., contains the information to be included in the definition line in sequence
headers. Modifiers are those included in the NCBI program SeqQuin.
Compose template - The procedure for composing a template for definition lines is straight forward:
Select the items you wish to include in the definition line from either of the two dropdown lists Project
variables or Modifiers. Click Insert to insert the item in the template field. The Project
variables are enclosed by <>'s (e.g. <My Project Variable) while the Modifiers are enclosed by
square brackets (e.g. [My Modifer]). Remember to click Close Entry when you have finished composing
a Modifier entry.
Format line - When the template is finished, click Format Line command button to see how the
information is displayed. In case you want to make changes, you can either delete the whole line by clicking
Clear Template or move the cursor to the point of correction and make the changes from the keyboard. Note
that Project variables enclosed by slashes are replaced by the relevant information extracted from the
project whereas Modifiers remain unaltered.
Build definition lines - When you are satisfied with the template, first click Unlock Line and then
the Build Def Lines command button to store FastA definition lines in all sequence headers.
As for all SEQtools functions changes and/or editorial changes are not stored until the project is saved. Closing the
project without saving it causes all changes to me cancled.
Template for a FastA definition line.
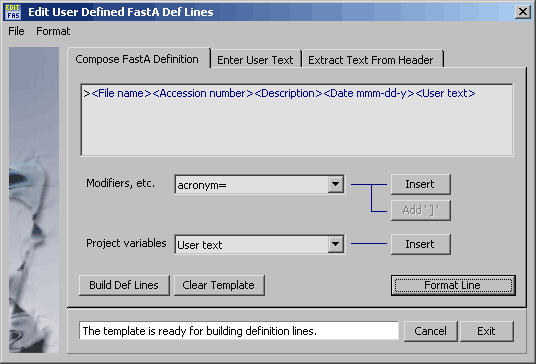
Formatted definition line. Note that empty project values are not included in the
definition lien.

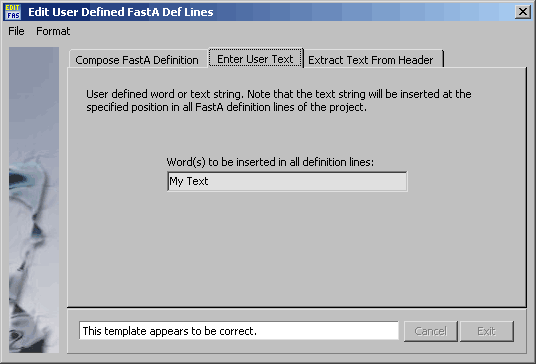
Form for entering User Text (the same for all definition lines).
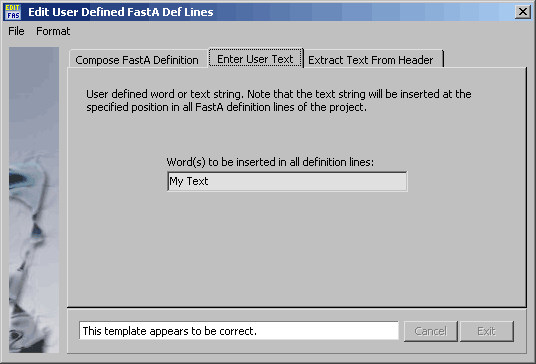
Form for extraction of header information to be included in the definition lines. The values depend on
header information for each sequence.
4.11.7 calculators
ASimple dna and math calculators.
![]()
4.11.7.1 DNA concentration calculator - Utility for calculation of molecular weight of DNA molecules.
Enter the number of base pairs and either the amount in nanograms or number of pmoles and press the Calculate
command button.
Single/double stranded DNA - Click SS to perform calculations on single stranded DNA or DS for double strand
calculations.
Nanograms - If the last entry is the number of nanograms, the corresponding number of pmoles is calculated.
Pmoles - If the number of pmoles is the last entry, the corresponding number of nanograms is calculated.
Output:
- Number of molecules
- Number of termini
- pmoles of termini
- Mw of DNA
Constants:
- Avogadros number = 6.022 x 10 e23 [molecules/mole]
- A = 331.20 [g/mole]
- C = 307.20 [g/mole]
- G = 347.20 [g/mole]
- T = 322.20 [g/mole]
- N = 326.95 [g/mole]
- Average mw of base pair = 2 x 326.95 - 2 x 18 = 617.9 [g/mole]

4.11.7.2 Math calculator - Simple mathematical calculator.
4.11.8 create files
Functions for creation and processing of different types of data files
4.11.8.1 Batch translation - Function for translation all nucleotide sequences in a project into protein. The
options for translation is shown in the screenshot of the form. The generated protein sequence files can be
saved after creation is accepted by the user.

4.11.8.2 EST submission to Genbank - A complex utility for creating a multi-sequence file for submission of
EST sequences to Genbank. The file includes the four TYPE files (Pub, Lib, Cont and EST)
required for submission. Each of the files is composed by adding information to one of four templates (Publication
Form, Library Form, Contact Form and EST sequence Form) containing all valid fields for that
particular TYPE file.
The EST template furthermore lets you insert information retrieved from the sequences loaded into the current SEQtools
project. Project variables are selected from a dropdown list and inserted by clicking the Insert
command button.
When each page of the template is finished, click the Verify Form command button to check if the entry is
correct and to convert it to the final TYPE text, leaving out empty fields. Obligatory fields are marked with
a leading underscore in the templates and fields for which information can be retrieved from the project
with a leading star (some fields in the EST template marked with a star are also obligatory).
When all four template tabs are finished, the submission file - consisting of a Publication record,
a Library record, a Contact record and one EST record for each sequence - is assembled
by clicking Build All. The submission file can be viewed by clicking View All and saved by clicking
Save.
When the file is saved, a submission Template file (with the extension tpl) is also saved in the same directory.
The *.tpl file can be used to retrieve the information included in submission and can be used for submitting
updates to the original submission.
Note that this requires that the exact same sequences with their headers are available to the function in the
current project.
After completing the submission file send it attached to an e-mail to Genbank
at batch_sub@ncbi.nlm.nih.gov. Further information can be obtained
from NCBI.
The EST Sequence Form of the submission utility. Values in fields in red are to be provided by project sequences and
their headers.

The final submission form, General information section.
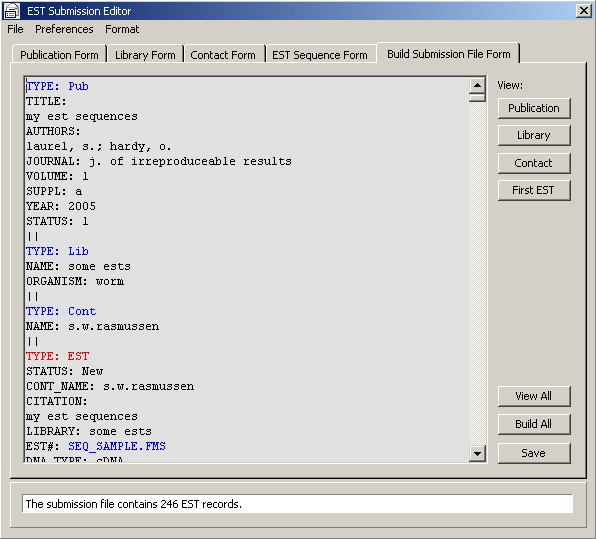
The final submission form, showing a part of the individual EST records section.

4.11.8.3 Sequence list, tab-delimited - This function is used to create and save a tab-delimited
list of project sequences. Clicking the Build command button causes the function to scan the displayed project
sequence list for available information. Available items are listed each with a checkbox. Information to be included
in the tab-delimited list is the simply checked in the list. Click Save when you are done.
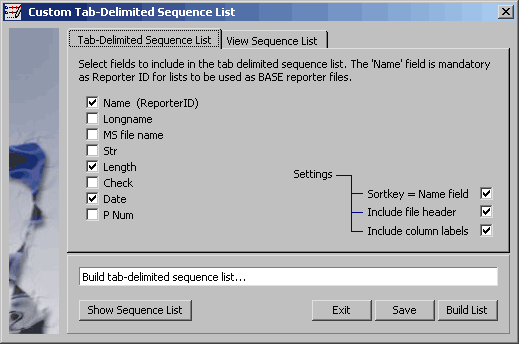
The selected items displayed in the View tab of the form.

4.11.8.4 Multi-record file parser - This user configurable text parser is described under
the Special menu.
© 2002-2010S.W. Rasmussen (revised: )